![]()
Hero of Hope
Carley Rios is a delightful 11 year old girl who has had a very difficult and complicated CF course. She was diagnosed as an infant and has had numerous and prolonged hospital admissions for pulmonary exacerbations. She also struggles with reflux, intermittent bouts of diabetes, weight gain, and small stature. Some people would find this difficult, but Carley takes on these challenges and remains forever optimistic about the future. For example, She was determined to become a "flyer" on her school cheer squad and, despite her physical limitations, she did!! She is an excellent student even though she misses many school days due to illness. Her aspiration is to attend college to become a pediatrician. Carley really wants to make a difference in the lives of other children who have CF.
Carley maintains a positive attitude while spending at least 3 hours per day on CF maintenance therapies. She also frequently requires home IV's. Carley faithfully follows this complex medical regimen without complaint and doesn't dwell on feeling sorry for herself, but continues to "live her life". She is kind, considerate, and supportive to family members and her many friends. She is an engaging, hopeful girl who can brighten a room with her smile.
Congratulations, Carley!
To nominate your own hero of hope or find out more about this program, go to pulmozyme.com and click on Heroes of Hope Living with CF.
To read more about Carley, check out this article in the Santa Cruz Sentinel
Cystic fibrosis lung infections
- What organisms cause lung infections in CF?
- Which ones can be spread from person to person?
- How can we prevent that spread?
Know Your Bugs
- Viruses
- Bacteria
- Fungus
Viruses
- Cause colds and flu
- Rhinovirus
- RSV
- Parainfluenza
- Influenza
- Can trigger CF exacerbation, hospitalization
- Commonly spread from person to person whether with CF or without CF
Pseudomonas aeruginosa
- Most common cause of lung infection in CF
- Is associated with worsening lung disease, especially "mucoid"
- Is getting more resistant to antibiotics
- Usually not spread from person to person,
- Rarely "bad bugs" are spread in CF
Burkholderia cepacia complex
- Rare in CF (less than 5% of patients)
- Can cause severe disease in about 25%
- Sometimes, patients have no ill effects
- Severe infections probably dependent on species of B. cepacia
- Naturally very resistant to antibiotics
- Frequently spread from person to person, especially "bad bugs"
Staphylococcus aureus
- Usually the first bacteria seen in young children with CF
- Can cause serious infections in healthy people
- Increasingly resistant to the usual antibiotics
- Can be carried even by healthy people and spread from person to person (CF and non-CF)
Haemophilus influenzae (H. flu)
- Same bacteria that can cause ear and sinus infections in non-CF children
- Hib vaccine prevents serious infections
- No effect of the vaccine in CF, the CF bug is not prevented by the vaccine
- Usually responsive to antibiotics
Other Bacteria
- Emerging, antibiotic resistant bacteria:
- Stenotrophomonas maltophilia
- Achromobacter xylosoxidans
- Seem to emerge with aggressive antibiotic treatment
- Probably not as problematic in CF as Pseudomonas and B. cepacia
- Not as easily spread as B. cepacia, Staph.
Non-tuberculous mycobacteria
- Relatives of TB (tuberculosis), but not nearly as dangerous to healthy people
- Some CF patients have problems with them, in others, harmless
- Require long treatment, multiple antibiotics
- Not spread from person to person
Aspergillus
- Can cause allergic problems, wheezing
- Allergic bronchopulmonary aspergillosis (ABPA)
- Can be treated with antifungal agents, steroids
- Can cause severe infections after transplant
- Other fungi rarely cause infections
- Comes from the environment, not spread from person to person
Wash Your Hands! How are germs spread?
- Droplet
- Coughing, sneezing, talking
- Can only go about 3 feet through the air
- Viruses and a few bacteria can be spread this way
- Direct contact
- Kissing, shaking hands
- Viruses and bacteria can be spread this way
- Indirect contact
- Shared eating utensils, respiratory therapy equipment, soiled tissues
- Viruses and bacteria can be spread this way
How can we prevent spread?
- Know your own bugs
- Recognize that things may have changed since your last culture
- Recognize that other CF patients may not know their bugs
- Comply with infection control recommendations to protect yourself and others
CFF Consensus conference on Infection Control
- Meeting in May 2001, Bethesda MD
- CF doctors, microbiologists, nurses, respiratory therapists, ethicists, lawyers, and CF patients all represented
- Doctors from Europe and Canada also there
- Tried to come to a consensus on the safest measures that will have the least effect on patients’ lives
Standard Precautions
- Recommendations from the CDC and HICPAC for all patients to prevent spread
- Hand hygiene (always!)
- Gloves when risk of direct or indirect contact
- Gowns to prevent soiling of clothes with contact or droplets
- Masks to prevent contamination of mucous membranes (eyes, nose, mouth) with droplets
To summarize…
- Know your bugs!
- Wash your hands!
- Become educated about how to protect yourself and others
Many patients and families are concerned about infection control and the measures taken at our CF Center to reduce the risk of transmission of resistant bacteria while continuing to foster a sense of community and support. Here are some relevant facts which may help explain our policies and procedures.Back to topicsFirst, it is essential to understand what we mean when we talk about resistance of bacteria to antibiotics. What does it really mean?
There are 4 categories of bacterial sensitivity to antibiotics:
- First, pansensitive - this means the bug is sensitive to all the antibiotics usually tested for potential treatment.
- Second, sensitive - the bug is sensitive to several potential antibiotics, but it may be resistant to others.
- Third, multiresistant - this is more complicated. According to the current definition, this means the bug is resistant to all antibiotics in two or more classes of antibiotics. Currently 3 classes of antibiotics are considered appropriate for treatment of Pseudomonas: certain beta-lactams, such as ceftazidime (Fortaz®); certain quinolones, such as ciprofloxacin (Cipro®); and aminoglycosides, such as tobramycin (Nebcin®, Tobi®).
- Fourth, panresistant - the bug is resistant to all tested antibiotics of all classes.
CASE STUDY
Here is an example of an antibiotic sensitivity report for fictional patient Jane Smith on a clinic visit expectorated sputum culture that grew three strains of Pseudomonas aeruginosa, one strain of Stenotrophomonas maltophilia, and Staphylococcus aureus. What is listed is the antibiotic sensitivity pattern for 1 of the 3 strains of Pseudomonas, i.e., just one of 5 lists of antibiotic sensitivities we would get from this single culture report on this patient on this day:
"3+ mucoid Pseudomonas aeruginosa Method Kirby Bauer Ticarcillin Resistant Piperacillin Resistant Ceftazidime Intermediate Ciprofloxacin Resistant Imipenem Resistant Gentamicin Resistant Tobramycin Resistant Amikacin Resistant Aztreonam Intermediate Cefepime Sensitive Meropenem Resistant
Tobramycin E test (not yet FDA approved) MIC 32 No interpretation" What is your categorization of this bug, and how should Jane Smith be approached regarding infection control on a clinic visit? You wouldn't be expected to know the answer without a lot more knowledge of how the tests are done and what they mean. Here are several points about this culture result:
- This bug is multiresistant. This is because it is resistant to all tested antibiotics in 2 out of the three 3 represented.
- The only quinolone tested (because it has the highest and only frequently present quinolone activity against Pseudomonas) is ciprofloxacin. Resistant.
- The three aminoglycosides tested are gentamicin, tobramycin, and amikacin. Resistant to all three.
- The rest of the drugs shown--total of 7--are beta-lactams. Of these 7 drugs, the bug is sensitive to just one, cefepime. This sensitivity keeps the bug from being called panresistant. But because it is multiresistant Jane will be asked to don a mask for her clinic visit, and if hospitalized she will have respiratory isolation procedures in place.
- Because it is multiresistant does it mean Jane is untreatable? No! If she is sick we can start cefepime (an iv-only drug). We would probably combine that with iv tobramycin, because even though the report says she is resistant to tobramycin there is synergy between these two antibiotic classes in attacking Pseudomonas.
- If Jane is a little sick, or her PFTs have dropped, we may put her on Tobi®. Why? Although the conventional test (Kirby Bauer disc diffusion) says her bug is resistant to tobramycin, the "E test" done at Stanford (not commonly done elsewhere) shows the bug can be killed by tobramycin if a concentration of 32 micrograms per milliliter [mcg/ml] can be achieved. It is difficult to do this by iv tobramycin without risking toxicity to kidneys or inner ear, but it is easy to do by having Jane inhale 300 milligrams of tobramcyin solution for inhalation-a Tobi® dose -- twice daily. This will produce a sputum level in the neighborhood of 1,000 mcg/ml, far above that needed to kill the bug, even after allowing for the fact that tobramycin can lose up to 90% of its activity in CF sputum.
THE CONCEPT OF RESISTANCE - A RED HERRING IN CF?
We have problems even defining and understanding what resistance means in the world of CF. The word resistance refers to lab tests done in a liquid suspension with a conventional set "dose" of bacteria and cutoffs between "sensitive" and "resistant" refer to levels of that antibiotic traditionally associated with curing systemic (i.e, blood) infections.We know in CF that the conventional term "resistance" is questionable clinically. For example, if your Pseudomonas is called "resistant" because tobramycin does not kill it at the conventional testing cutoff level of 8 mcg/ml, this is not very relevant in a disease where the bug is living in the mucus rather than tissue or blood, and we can deliver 1,000 mcg/ml Tobi® to the mucus by aerosol. Moreover, the way the bug lives in your lung is as a mucoid biofilm-a gigantic communal mass of bacteria (up to one billion bacteria in one gram of sputum!) stuck together by a slimy material called alginate or mucoid exopolysaccharide. This is much different than the way lab antibiotic testing is done with individual bugs swimming around freely in liquid suspension.
INFECTION CONTROL
We rely on universal direct and indirect contact precautions for ALL patients to reduce transmission of bacteria. The simplest way to describe this is to imagine a three foot zone around your body that should ideally not be entered by another patient (unless you live with them), and no shared object use without proper cleaning in between. All contacts by caregivers should be preceded by handwashing and cleaning of multi-use instruments such as stethoscopes.We are concerned about acquisition and spread of Pseudomonas, and the infection control measures are designed to reduce the chance of spread. For multiresistant and panresistant bugs, we add restrictions on the patient such as use of mask when coming to clinic, and isolation when hospitalized. This is done to reduce the chance of transmission, not because the patient is sicker! The mask is an additional level of precaution but contact precautions are the key element for ALL patients.
In the near future the CFF Consensus Conference report on Infection Control will be published and available for detailed reading.
REPORTING CULTURE RESULTS TO PATIENTS
Ideally, we would like to track levels of resistance in all cultures and inform all patients promptly. In practice, this is a very difficult task. In our center we see over 200 patients a year, on average 4 times, yielding at least 800 "surveillance" bacterial cultures alone. Add to this cultures obtained when the patient is sick, and when hospitalized. Each specimen may contain multiple bacterial strains that are each tested for antibiotic sensitivities. And this is just for bacteria; often further cultures are also obtained for molds, mycobacteria, and so on. Finally, many other tests (bloodwork, x-rays, PFTs) are also being monitored.Given our staff resources we have in the past focused on the priority of promptly informing people who are culture negative for Pseudomonas when they pick up Pseudomonas because we want to try to eradicate it early. Later on this becomes impossible, so that most people who have years of carrying mucoid Pseudomonas cannot achieve eradication by any known treatment. We do not routinely inform patients of each culture result in that instance. Besides the impracticality of the numbers, we know that typically a strain may go from "resistant" to "sensitive" and back and forth over time, confusing many people and raising needless anxiety. Instead, we focus on dealing with the situation at each clinic visit as well as of course during hospitalizations.
The infection control guidelines currently being prepared for publication do not distinguish between patients with sensitive or resistant Pseudomonas outside health care settings, but rather recommend a uniform set of infection control behaviors for all persons with CF. Thus, we have no specific reason to routinely inform patients with long-term Pseudomonas carriage of culture-to-culture changes in their resistance pattern with regard to their outside the health care system interactions, but urge adherence to the general recommendations. Cultures are checked before the next clinic visit so we can institute appropriate infection control for that visit.
Recently, our staff met and decided to go beyond current standard practice and monitor sensitivity results for long-term Pseudomonas patients, and send a letter out informing patients with resistant organisms of the mask requirement for clinic. We hope this system minimizes surprises about our infection control measures in the clinic and hospital. It is critical to remember that a "resistant" bug report does not and should not be taken as a bad thing in and of itself, and that our main interest as to timing use of this information focuses on our health-care facility setting, and early monitoring of new Pseudomonas. Now, in addition, we will attempt to track long-term carriers and provide "early warning" infection control letters.
Antibiotics are essential part of treatment for cystic fibrosis lung disease. Most patients receive numerous courses of oral, iv or inhaled antibiotics for symptoms and many patients are maintained on long-term "suppressive" regimes of oral or inhaled antibiotics. It is not surprising that given this enormous exposure a substantial problem with allergic reactions exists. Most reactions and reports of sensitization have been to the class of antibiotics derived from penicillin that are effective against Pseudomonas aeruginosa, the major bacterial infection in CF. These are called anti-pseudomonal b-lactam antibiotics [BLA]. Allergy to aminoglycosides such as tobramcyin and quinolones such as ciprofloxacin are very rare.A general principle of allergy is that reactions are more likely the more closely related two molecules are. Within the BLA family, there are 4 subfamilies chemically related by a core structure called the b-lactam nucleus but quite different in their side chains. The BLA subfamilies and the drugs used in CF are as follows:
It turns out that most BLA allergic reactions occur due to allergic or IgE antibodies being formed against the side chains, so most reactions occur upon re-exposure to the same drug, or to a closely related drug in the same subfamily with a similar side chain structure. The chances of a reaction diminish as the structure becomes more dissimilar.
- Penicillins
- Carboxypenicillins
Ticarcillin (Ticar, Timentin)- Acylaminopenicillins
Piperacillin (Pipracil, Zosyn)- Carbapenems
- Imipenem (Primaxin)
- Meropenem (Merrem)
- Monbactams
- Aztreonam (Azactam)
- Cephalosporins
- Ceftazidime (Fortaz, Tazicef)
- Cefepime (Maxipime)
Allergic reactions to BLA occur in 1-10% of subjects during a given course of treatment, with the variation in incidence dependent upon several factors such as the subject population, drugs involved, and ascertainment methods. The vast majority of these are limited to skin or underlying tissues, with only 2-10% involving life-threatening respiratory or cardiovascular reactions. Up to 9% of the life-threatening reactions may result in death, yielding a final fatality rate estimated at about 1 in 50,000 treatment courses.
Most of the information in the medical literature is based on allergy to penicillin itself, not the many derivatives such as those used against Pseudomonas. Ten to 73% of patients giving a history of penicillin allergy react on allergy skin testing to penicillin-derived chemicals or reagents, depending upon time elapsed since the reaction, nature of the reaction, age of subject, and other factors. Evaluation of penicillin allergy with just two commercially available reagents, penicillin and its major metabolite penicilloyl-polylysine [Pre-Pen®], detects 65-93% of allergic individuals. The remainder of allergic subjects are reactive only to minor penicillin metabolites which are unstable and not commercially available. Unfortunately, those sensitized to these minor metabolites are at higher risk of more serious reactions if re-exposed to penicillin.
For people sensitized to BLAs other than penicillin, skin testing and prediction of subsequent clinical reactivity becomes much more difficult. Major and minor metabolites of these drugs are not commercially available and are not well characterized. Cross-reaction studies between various BLAs yield varying results and clinical responses are not predictable from skin test responses. Thus, the safest course is usually to avoid BLAs and choose an alternate class of drug when possible. However, treatment of Pseudomonas aeruginosa infection rarely offers this choice; usually, a BLA is essential.
Studies of BLA allergy in CF have been reported since 1970, including a number studies from our CF Center over the last 15 years. In general, these show BLA allergy rates of about 15-25% in CF patients and 5-10% of iv BLA treatment courses. These reactions are usually in-hospital, physician-observed, well-characterized and documented. Reactions are usually seen in older patients with multiple prior exposures. Reactions always include the skin (itching, redness, hives, swelling) while respiratory involvement occurs in only a small minority. However these can be quite severe with bronchospasm or largyngeal edema. Cardiovascular problems are very rare.
Unusual cases of patients with reactions to several classes of antibiotics, not just BLA, have been reported also.
Our studies using skin testing and laboratory immunological assays suggest that BLA allergy in CF is mainly drug-specific. This situation differs from other penicillin allergic patients in the literature, whose allergy appears mainly directed to the core BLA nucleus. It is clear that cross-reactivity between various BLA occurs, and that any BLA may cause an allergic reaction. It is not currently possible to predict which CF patients will become sensitized, although multiple courses, high doses, and the intravenous route of drug exposure are key elements in sensitization. Patients may become sensitized during or between any given course of antibiotic therapy.
Treatment
The main treatment strategy in CF is called desensitization. Desensitization is the administration of gradually increasing doses of a drug under close observation for adverse effects. Desensitization has been performed safely in pregnant women, critically ill patients, and patients with serious but not life-threatening infections. How desensitization works is incompletely understood.
Desensitization may be performed orally with oral drugs, but most BLA used in CF are given only by the iv route, so most desensitization procedures are done intravenously. In addition, only the iv route allows control of the dose and rapidity by which the drug enters the bloodstream. During iv desensitization, the earliest signs of a reaction can be at least in part immediately managed by slowing or stopping the infusion. If the reaction history was severe pretreatment with Benadryl and sometimes steroids may be included. We have performed many hundreds of iv desensitization for BLA allergy in CF since 1983. Skin rashes occur in 25-30% of cases, often during or just after the last and highest dose. Mild reactions are treated with slowed infusion rates and iv Benadryl. More serious reactions occur in 1-2% of cases and require immediate management and discontinuation of desensitization. In virtually all of these cases, iv desensitization with another BLA has been successfully performed 1-2 days after the patient has been stabilized. Desensitization relies on staying on the prescribed drug schedule; missed doses increase the possibility of a reaction.
While it is possible to maintain desensitization by indefinitely staying on the drug in question, in most cases this is impractical in CF. Instead, we rely on history (identifying prior drug reactions) and repeat desensitization if a BLA causing a reaction needs to be given again. In many instances, an alternative BLA can be chosen instead. Studies show the likelihood of a cross-reactive allergic reaction is in the range of 2-5% if a different BLA subfamily drug is used (e.g., you reacted to piperacillin and we then use ceftazidime). Often in this situation we will give a test dose (1/10 or 1/100 of the full dose) first, and then the full dose if no symptoms occur.
Delayed reactions. Delayed reactions take days or weeks to occur and are not life-threatening, but can be quite troubling. They usually consist of fever, malaise, muscle aches, non-itchy flat irregular red rashes, and sore sometimes swollen joints. Rarely enlarged glands or internal organs, or more severe skin rashes may occur. Delayed reactions are more common with piperacillin than other BLAs. Besides stopping the drug, steroids are often given. Desensitization does not prevent delayed reactions if the same drug is given again.
What You Can Do
Know your drug allergy history! Record the name of the drug, when the reaction occurred and what happened. Do NOT assume the medical record is foolproof - it is not! YOU should take charge of your medical history and make sure you are not exposed to a BLA you reacted to in the past without proper evaluation and a plan.
Infant Pulmonary Function Tests
by Colleen Dunn RCP
Pulmonary function tests (PFTs) are a group of breathing tests designed to determine how healthy someone's lungs are. As many of you know, when your child becomes 5 or 6 he or she is introduced to spirometry (basic pulmonary function test). The reason for waiting until the child reaches this age is to achieve the cooperation of the child and to be able to obtain meaningful information.
Infant pulmonary function tests are performed when your doctor would like to assess your infant or toddlers' lung function, but they are too young to do spirometry. We now have a NEW infant pulmonary function system at LPCH that will enable us to measure more accurately lung function in very small children. Dr. Conrad, one of our pulmonary specialists, and her associates sedate the children in order to help them relax and fall asleep. Due to small children's inability to cooperate, they must sleep soundly so that they don't wake up during the test. A mask is placed gently on the face during the measurements. The testing period normally takes about 20 minutes after the child has fallen asleep. We are able measure how big each breath is. We can mimic the FVC (forced vital capacity) of the spirometry test by gently "hugging" your child's chest. We are able also to measure the amount of air in the lungs that no one can blow out (the remaining air). It can be determined if your child's lungs act like someone with asthma.
Infant pulmonary testing is useful and relatively easy to accomplish in children (as early as 6 months of age) to obtain a baseline of your child's lung function. The study can be performed again months later to compare the results to prior studies. Results can be followed over time to see how well your child is doing, (for example: how well he is responding to current therapies) just as the results of routine spirometry are used by your doctor during regular clinic visits.

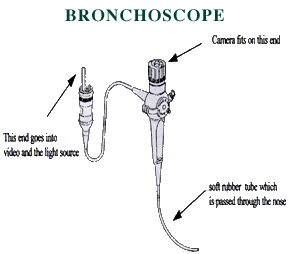
Bronchoscopy
by Carol Conrad, MD, Stanford CF Center
Your doctor may advise you that your child should undergo a procedure called a bronchoscopy. A bronchoscopy is a procedure that allows us to look at the airways, or the tracheobronchial tree, through a special sort of “scope”, called a bronchoscope. With the bronchoscope, we can evaluate the respiratory system, including the voice box (larynx), the wind pipe (trachea), and the airways (bronchi) for evidence of any abnormality or infection.
Why is a bronchoscopy done?
Sometimes it is important to watch the movement of these structures during the different stages of the breathing cycle. At times it is important for diagnostic purposes to obtain some of the fluids in the lung through the bronchoscope for evaluation in the laboratory. The scope has a special channel through which we can vacuum some of the liquid lining from the airways. A bronchoscopy allows a physician to perform diagnostic tests, to obtain specimens for biopsy and culture, or to remove foreign objects that children might have inhaled.
What is a bronchoscope?
The bronchoscope is a soft (flexible) tube with an outer diameter as small as 2.2 mm and there are larger sizes for use with larger children and adults. The scope can be easily passed through the nose or the mouth. The flexible tube actually contains a fiber-optic system which attaches to a video camera and a source of light. The light travels through the scope and lights up the inside of the airway. The image seen at the tip of the scope is transmitted back again through the fiber-optic system to the video camera. We can also take photographs through the scope. What information do you get from a bronchoscopy?
A bronchoscopy enables the physician to see and test the material that is deep in a patient's lungs. Sometimes bacteria lodge deep in the lungs, and it can be difficult to find or identify them. This material can be washed out by washing the airways with saline. This is referred to as a bronchoalveolar lavage (BAL). The physician injects a small amount of saline through the bronchoscope into the airways and then sucks it back through the suction port of the bronchoscope. The fluid obtained contains saline plus secretions from the lung: bacteria (if present), cells, etc. This sample is sent to the laboratory for testing.
How is a bronchoscopy done?
A bronchoscopy is done in a special room in the outpatient ambulatory procedure unit at the Packard Children's Hospital. Typically, a bronchoscopy is performed with the patient sleeping, but breathing on their own. The patient is given medications to produce sedation (a sleepy state). At times, biopsies, or very small pieces are obtained from the lungs, and this may require the use of general anesthesia, but most bronchoscopies are done under sedation. The patient is arousable from this sleep and is able to cough, sneeze, or may try to speak if directed. We keep a close watch on the patients with monitors that continuously check the patient's vital signs such as heart rate, blood pressure, and blood oxygen levels (oximetry).
After the patient is asleep, the next step is to numb the nose and back of the throat. Numbing drops (Lidocaine) are sprayed into the nose. Later on during the procedure, this medicine can be applied to the airways though the bronchoscope to numb other structures such as the larynx, trachea, and bronchi as they are encountered. The Lidocaine prevents irritation from the scope to the airways, and can minimize coughing and sneezes. After the numbing medicine is applied, the bronchoscope is inserted through one nostril, and the procedure begins.
How long will this take?
The patient usually comes to the outpatient surgical center an hour or so before the scheduled bronchoscopy begins. This allows time to fill out insurance and medical forms, and in the pre-operative unit, vital signs are recorded, and a brief physical exam is done. At about this time, an IV line is placed by the nurses. The actual bronchoscopy takes about 45 minutes. After the procedure is done, the patient recovers in the post-anesthesia care unit until he/she is awake and is able to go home. The medicines used for sedation are considered to be “short-acting”, but parents often report that the baby sleeps through much of the rest of the day. The entire procedure takes about 4 hours in the hospital.
What are the risks of the procedure?
Flexible fiberoptic bronchoscopy is generally very well-tolerated and safe, even in very young infants. However, there are certain complications that are associated with flexible bronchoscopy. The most common problem is a fever that occurs after the procedure. This is usually a low grade fever (rarely over 101°) and usually responds to acetaminophen (Tylenol, Tempra, or other brands). Your child may experience some mild bleeding from the nose. This responds to pressure and is rarely encountered in children who are not predisposed to bleeding problems. In patients who are having a biopsy performed, a pneumothorax is a concern. This results in the collection of air around the lung and may result in collapse of a lung. A tube may need to be placed to evacuate the air and allow the lung to re-expand. Your child may require supplemental oxygen during and after the procedure. If he/she is having respiratory difficulties, we may need to place a temporary breathing tube in your child's airway and breathe for him or her. Finally, oversedation may necessitate a longer recovery period for your child and in rare instances, an overnight hospitalization.
Respiratory Treatments
- Airway Clearance
by Kristin Shelton, RRT- Pulmonary Function Testing
by Kristin Shelton, RRT- Nebulizers and Compressors
by Deborah Gilley RCP and Kristin Shelton RRT- Choosing a Nebulizer
reprinted from the Winter 2002 CF Center Newsletter
Airway Clearance - Something for Everyone
by Kristin Shelton, RRT
- Chest Physiotherapy (CPT)
- Positive Expiratory Pressure (PEP)
- Vibratory Positive Expiratory Pressure (Flutter® , Acapella®)
- High Frequency Chest Wall Oscillator (Vest)
- Intrapulmonary Percussive Ventilation (IPV)
- Active Cycle of Breathing
- Autogenic Drainage
- Long Term Considerations
Chest physiotherapy (CPT) is the traditional means of airway clearance in CF. It uses postural drainage in various positions, percussion, vibration, deep breathing, and coughing to loosen and move secretions out of the lungs. The treatment time including an aerosol before is about 45 minutes.Back to top
Benefits: CPT is a time proven, widely accepted method for airway clearance. It generally requires a minimal amount of effort on the part of the patient, but is much more effective when coughing and deep breathing accompany it. CPT does not require the purchase of specialized equipment. If a tilt table is not available, CPT can be performed on a bed or sofa with the use of pillows or wedges. CPT also promotes a special time together for the caregiver and the patient.
Disadvantages: Because CPT requires the assistance of a caregiver, it becomes necessary to rely on another person whenever airway clearance is needed.The caregiver performing CPT can be at risk for repetitive strain injuries.
Cost: If you have to hire someone to assist you, the cost can be quite prohibitive. Hiring a professional to perform CPT is approximately $ 150 each treatment. At 2 treatments per day the cost per year can be $ 108,000. The overall cost of years of expensive airway clearance can deplete your insurance lifetime maximum quite rapidly.
Another consideration these days is the lack of availability of care providers. If you are dependent on a professional to receive regular routine airway clearance, and for reason the caregiver cannot come (staffing, reimbursement, or location, etc.), it is very important to have an alternative, effective form of airway clearance available to you.
Practicality: It is not always convenient to perform CPT just anywhere. It requires space and planning to make CPT portable. Therefore it is necessary to look at some alternatives.
Back
PEP is a technique that uses a hand held device which can be used with a nebulizer attached. It has a restricted orifice. When exhaled into, this creates pressure in the lungs. This pressure allows air to enter behind areas of mucus obstruction and keeps the airways open during exhalation. As you exhale, mucus moves towards the larger airways, so it can be more easily coughed up with the huff technique. PEP can be taught to children as young as 5 years, and can be passively given to infants via a mask. The treatment time is about 20 minutes.
PEP Benefits: PEP can be performed without the help of a caregiver, but does require supervision in young children. PEP can be performed with the nebulizer attached (this saves time). It is effective without need for postural drainage (head down) positions. The patient can also be participating in some other activity during the PEP treatment which might increase compliance. PEP is easy to learn, can be performed almost anywhere, does not require bulky equipment, and is easy to clean.
Disadvantages: PEP is ineffective when it is performed incorrectly. Therefore, there is a need for supervision when children are performing PEP.
Cost:$ 30 each
Practicality: The PEP device is inexpensive, it can be performed without a caregiver, and it is small and portable.
BackVibratory Positive Expiratory Pressure (Flutter®, Acapella®)
Vibratory positive expiratory pressure is a hand held device. Exhaling into this device results in oscillations of pressure and airflow which vibrate the airway walls (loosening mucus), helps hold the airway open (which allows air to get behind secretions and keeps the airways open during exhalation). It speeds up airflow helping mucus move up to the larger airways where it can be more easily coughed up. Vibratory PEP can be taught to children as young as 2 years old by mask, and to ages 5 and up via mouthpiece. Treatment time is about 20 minutes.
Benefits: Flutter and Acapella are easy to learn, and very portable. This form of airway clearance can be performed without the assistance of a caregiver. An added advantage of the Acapella brand is the ability to attach and use a nebulizer during the airway clearance. The Acapella also has an adjustable expiratory pressure dial, and unlike the Flutter it can be used at any angle.
Disadvantages: Like the PEP, the Flutter and Acapella are ineffective if used incorrectly and require supervision.
Cost: $44.00
Practicality: Because they are inexpensive, portable, and easy to use, the Acapella and Flutter are very practical.
BackThe Vest
The High frequency chest wall oscillator is a large device in which a nonstretchable vest is worn. The vest is connected to an air pulse delivery system by way of tubing. A nebulizer is used at the same time. The air-pulse generator delivers high frequency vibrations which then squeeze and vibrate the chest wall. These vibrations, along with an increase in airflow, help loosen mucus from the lungs.Children as young as 3 years are able to use the vest. A treatment lasts 20-30mins (depending on the size of the chest).
Benefits: The vest is done sitting upright. This therapy can be done independently. The vest is designed to provide a lifetime of use (the vests are adjustable and multiple sizes are available). The vest is easy to use. It does not require a special technique or effort to be done correctly.
Disadvantages: The vest is large and heavy. It can be transported, but not conveniently. The vest is noisy and far from discrete, which can lead to a lack of compliance.
Cost: $16,000
Practicality: The vest is adjustable, easily repairable and made to last a lifetime. Because there is no caregiver required, the vest is very practical.
BackIntrapulmonary Percussive Ventilation.
The IPV is a pneumatic (air driven) device that delivers both continuous airway pressure and mini bursts of air. At the same time the IPV delivers a dense aerosol.The combination allows air to enter behind mucus blockage, and vibration to dislodge mucus from the airway walls so it can be more easily coughed up. IPV instruction can begin at age 12 or so, and it can be used in younger patients via mask, but this method is not well tolerated. The treatment time is about 20 minutes.
Benefits: The IPV is a device that does not require a caregiver, and has the ability to give the aerosol and airway clearance at the same time.The home model of the IPV is electrically operated and portable (but is quite heavy).
Disadvantages:The IPV is quite vigorous and some pts do not tolerate this. The IPV is ineffective if performed incorrectly and can be somewhat difficult to learn. The IPV is rather noisy because of the large compressor required to drive the device. The Percussionaire brand IPV has many rather delicate, expensive parts, and can require frequent maintenance. The Vortran type IPV has a fully replaceable percussion device, but it is not very adjustable (in regards to the speed and strength of vibration).
Cost: The Percussionaire brand is $8,500. The Vortran brand is $1,000.
Practicality: The IPV is a long term, fairly reliable form of airway clearance. It is expensive and can be delicate, but has the convenience of not requiring a caregiver.
BackActive Cycle of Breathing
Active cycle of breathing is a series of breathing techniques, consisting of thoracic expansion exercises (deep breathing), breathing control (using the diaphragm), and the forced expiration technique (huff). These breathing cycles are performed in various positions of drainage similar to CPT positions but without the percussion. This can be taught at about the age of 8 years. Treatment time, including an aerosol before, is about 45 minutes.
Benefits:Active cycle of breathing is fairly easy to learn, and does not require the assistance of a caregiver. It does not require any specialized or expensive equipment.
Disadvantages: Like CPT, ACB requires a surface on which to position the body in postural drainage positions.
Cost: The only cost is the initial training.
Practicality: Because there is no cost, no need for a caregiver, and no specialized equipment, ACB can be very practical.
BackAutogenic Drainage
Autogenic Drainage is a breathing technique which involves 3 phases of breathing levels:A huff maneuver is used to cough up secretions, and then the phases of breathing are repeated again. The earliest age for learning AD is about 12 years. The treatment time, including a nebulizer before, is about 35-50 minutes.
- The first phase is the unsticking phase which is inhalation and exhalation of small amounts of air.
- Phase two is the collection phase where medium sized breaths are inhaled and exhaled.
- Phase three is the evacuation phase where large amounts of air are inhaled and exhaled.
Benefits: Autogenic Drainage can be performed almost anywhere at any time. AD does not require the assistance of a caregiver. AD gives a patient very good control of their breathing and a heightened awareness of the lungs and their condition. This awareness and control becomes significantly helpful later in the course of CF when shortness of breath becomes an issue.
Disadvantages: AD is difficult to learn and takes a lot of practice to become proficient. To be an effective form of airway clearance it also requires a good deal of self-motivation, concentration, and discipline.
Cost: There is no cost associated with Autogenic Drainage except for the cost of the initial training.
Practicality:AD is very widely used in Europe and parts of Canada. It is definitely versatile and convenient, but because of the commitment and discipline required for proficiency, it hasn’t become common in the U.S.
BackLong Term Considerations
When deciding on a form of airway clearance it is important to consider many factors.As children mature and move through adolescence to adulthood, there should be a degree of independence gained. When it comes to airway clearance, it is very important to choose a form of airway clearance that will afford you the opportunity for independence that you desire and deserve. It is important to take responsibility for your own healthcare, and that includes airway clearance.
- How much time are you willing to, and realistically able to devote to airway clearance?
- If you choose a form of airway clearance that requires the help of a caregiver, do you have a person available to assist you unconditionally?
- Have you had a trial period using the chosen airway clearance to make sure it is effective and well tolerated by you before time and money is invested in its purchase?
- What are your plans for the future... If you plan to go to college, is your choice for airway clearance something portable, discrete, and realistic?
- If you need to hire a professional organization such as home health care, what is the long term cost?
- How realistic is it to expect the company to continue to supply expensive treatments to you despite a usually poor financial reimbursement?
- How much of a drain is occurring to your insurance lifetime maximum? (you may want to conserve this valuable resource for future needs such as hospitalization or transplant).
Studies considering the effectiveness of the various airway clearance techniques have shown good results with all the various methods. However, consideration of the right airway clearance must include
- Physician recommendation
- Acceptance and feasibility for the patient
- Compliance with the chosen method
Back to Respiratory Treatments
Everything You Always Wanted to Know About Pulmonary Function Testing But Were Afraid To Ask
by Kristin Shelton, RCPWhat are PFT's?
Pulmonary Function Tests (PFT's) evaluate how well the lungs work. They measure:
As Cystic Fibrosis progresses and changes occur in the lungs, the relationships between volumes and capacities also change. Lung volume is the amount of space in the lungs. Lung capacity is the maximum amount of air that can be contained in the lungs.
- How much air volume can be moved in and out of the lungs (measured in liters)
- How fast the air in the lungs can be moved in and out (measured in liters per second)
- How stiff are the lungs and chest wall
- How the lungs respond to airway clearance techniques (flutter, vest, percussion)
Why we use PFT's in CF clinic:
Often, seeing the results of your PFT's can be confusing or upsetting. Hopefully with some explanation of the different abbreviations and terms frequently included in PFT reports, the understanding of your own test results will become clear.
- To keep track of the progression of pulmonary disease.
- To keep track of the effectiveness of treatment.
Pulmonary Function Tests
The term pulmonary function test is a general term. Just as there are many blood tests, there are many pulmonary function tests. Each test gives us different information. These are some common pulmonary function tests:
Spirometry
The most basic pulmonary function test is called spirometry. This is the test that is carried out on a routine basis and at nearly every clinic visit. This test gives a series of values which allows comparison of current lung function with lung function in the past. Following spirometry closely allows early intervention if lung function is falling. Please note: if you carry out spirometry testing several times during one day or once a day for several days in a row there will be changes of a few percent in the various numbers measured. These small changes have very little clinical meaning. Larger changes over a longer time (e.g. the time between clinic visits) may indicate a need for adjustments in your therapy. Simple spirometry is an important test, which allows the C.F. team to evaluate the disease process, determine the effects of illness on pulmonary function, and look for a response to changes in therapy.
Simple spirometry includes:
FVC: Forced Vital Capacity After taking in the deepest possible breath, this is the volume of air that can be forcibly and maximally exhaled out of the lungs until no more can be exhaled. FVC can be reduced in C.F. because of mucus plugging and airway constriction.
FEV1: Forced Expiratory Volume after one second This is the measurement of the maximum amount of air which can be forcibly exhaled from the lungs in the first second of a forced expiratory maneuver. A healthy person can exhale 75-85% of the FVC in one second. This can be decreased because of narrowing of the airways, airways becoming obstructed with mucus, or because of swelling.
FEV1/FVC: The ratio of FEV1 to FVC expressed as a percentage This indicates what percentage of the total FVC was expelled from the lungs during the first second of forced exhalation. FEV1/FVC can be reduced in C.F. because of blockage caused by mucus.
FEF 25-75%:Forced Expiratory Flow from 25-75% This is the average rate of air flow during the middle half of a forced exhalation. This shows the condition of medium-sized and small airways. In early stages of an obstructive disease like C.F. decreased flow rates are common.
FEF Max: Forced Expiratory Flow at Maximum effort (or Peak Flow) This is the maximum flow rate attained during a forced exhalation. This is a good measure of a patient's effort. The peak flow can be reduced in C.F. when there is severe blockage of the small airways but is often seen with large airway narrowing (such as Asthma).
SVC: Slow Vital Capacity This is the volume of air that can be measured in a slow, complete exhalation after a maximal inspiration, without forced or rapid effort.Full Pulmonary Function Testing includes all of the above measurements plus:
TLC: Total Lung Capacity is the volume of air contained in the lungs after a maximal inspiration. In C.F. the TLC can be larger than predicted because of air trapping.
FRC: Functional Residual Capacity is the volume of air remaining in the lungs at the end of a normal quiet expiration.
RV: Residual Volume is the volume of air remaining in the lungs at the end of a maximal expiration. Increase of RV (in other words: even after a maximal expiration, the lungs still contain an abnormally large amount of air) in C.F. can be caused by air trapping.
TGV: Thoracic Gas Volume is the volume of air contained in the thorax (chest cavity). This is a more accurate measure of the FRC, because it measures all gas within the thorax, even trapped gas. This test is performed in the Body Plethysmograph, better known as ‘the box’.
MIP: Maximal Inspiratory Pressure This is a measurement of inspiratory muscle strength.
MEP: Maximal Expiratory Pressure This is a measurement of expiratory muscle strength.Other common measurements include:
Pulse Oximetry also called ‘saturation’ or SpO2, is a measure of how well a person's blood is able to carry oxygen. There is a predictable correlation between the oxygen saturation level and the actual amount of oxygen in the blood (as would be measured by an arterial blood gas). Normal ranges are between 95-100%. A measure of your oxygen saturation can be done easily and painlessly with a clip or Band-Aid type probe that is placed on the finger. This probe has a light that shines through one side of your finger and a receiver that measures the light that comes out through the other side of your finger. The amount of light received indicates how much oxygen your blood is carrying. A more accurate measure of the oxygen in the blood is obtained from an arterial blood gas.ABG: Arterial Blood Gas is a small sampling of blood drawn directly out of an artery. The artery that is sampled most often is the radial artery in your wrist, (the same one you can feel when having your pulse taken) This test allows a very accurate measurement of the following values:
- pH: This is a measurement of your blood's acid-base balance. Normal ranges are 7.35-7.45. Values less than 7.4 are considered to be more acidic, while values greater than 7.4 are considered more basic. If there is a problem with breathing or metabolism, there will be an effect on the pH.
- PaCO2: This value indicates how effectively your lungs are able to rid themselves of CO2. The normal range for PaCO2 is 35-45mmHg. Values greater than 45mmHg usually indicate that the lungs are not able to work well enough to rid themselves of the CO2.
- PaO2: This is a measure of the amount of oxygen present in your arterial blood. The normal range for PaO2 is generally between 75-80mmHg depending on your age. If this value is found to be less than 55-60mmHg, your physician may recommend supplemental oxygen.
- HCO3: Bicarbonate: Bicarbonate is the body's natural way of buffering the blood. The kidneys regulate the bicarbonate level in the blood. The normal range for HCO3 is 22-28meq/L. Elevated bicarbonate can occur when there is an abundance of CO2 because the lungs are not working properly.
Pulmonary Function Testing can be a complicated subject to fully understand. Your Respiratory Therapist is an excellent resource for any questions you might have on this important topic.
Back to Respiratory Treatments
PLEASE NOTE: A national consensus conference on infection control has resulted in some changes in nebulizer care. Please check our education section on Nebulizers for current information.
Nebulizers and Compressors
by Deborah Gilley RCP Clinical Specialist Respiratory Care
and Kristin Shelton RCP Respiratory Care Coordinator for the CF centerNebulizers and compressors are devices for delivering aerosolized medication. The nebulizer changes liquid medicine into a mist so it can be inhaled into the lungs. Driven by a compressed air machine, the nebulizer consists of a cup which holds the medicine, a mouthpiece attached to a T-shaped part or a mask, and thin, plastic tubing to connect to the compressor. Nebulizers and compressors are an integral part of the treatment plan for cystic fibrosis.
NEBULIZER CARE CAN BE SUMMARIZED IN JUST THREE STEPS:
- Clean it at least every 24 hours
- Disinfect it regularly
- And most important of all BE SURE YOUR EQUIPMENT IS DRY BEFORE USE
If you are using a nebulizer every day, consider having three nebulizer sets so that you always have one that is clean, dry, and ready to go. Non-disposable equipment is stronger and lasts longer than the disposable variety. Some non-disposables may be dishwasher safe. The disposable equipment usually lasts for about one to two months, although some may last longer. After repeated washing they may not work as well, making your treatments take a lot longer or possibly affecting the amount of medication that is delivered. Also if you ever break or crack your nebulizer, you should replace it with a new one.
There are many types of nebulizers to make them appeal to consumers, such as ultrasonic, jet, and sidestream. When choosing a nebulizer, make sure the particle size (of delivered drug) is within the recommended range of 0.8 to 3.0 microns. And be certain that the medication to be delivered is intended for use in the nebulizer you choose ( for example... Pulmozyme cannot be delivered by ultrasonic nebulizer because heat is generated during the operation of the ultrasonic nebulizer which causes the Pulmozyme to become unstable).
PLAN AHEAD:
In this era of potential rolling electrical brown-outs, or strong winter storms causing power outages, you should have an alternate plan for medication administration.
- One would be to use a battery operated compressor, such as the one that works from a car cigarette lighter.
- There are also some compressors that operate using batteries. Consider checking with your physician or insurance provider for authorization for a second "back up" compressor or give it consideration when you need a new one.
- Another alternate plan would be to use a metered dose inhaler (MDI) or a dry powder inhaler.
MDI VS NEBULIZER:
- Even though the MDI takes only a few minutes, as opposed to the 20 minutes for the nebulizer, it will still take about 20 minutes before you start feeling the peak effect of the medication after using the MDI.
- An MDI and a nebulizer deliver the same medication, but the doses of medication will be different. Check with your Doctor or nurse for the correct dosage.
- Unfortunately, not all medications can be used in an MDI. Mucolytics and antibiotics, for example, are only available as liquids, and so must be nebulized.
TECHNIQUE IS EVERYTHING
All equipment is technique dependent. Pay attention to how you use your equipment for the best possible results. Ask your health care provider to teach you the correct technique, especially if you get something new.
BAFFLE THOSE BUGS
Bacteria thrive in moist environments (like the lungs). They usually do not survive on a dry surface, so be sure your equipment , no matter what kind, is DRY before initial use.
CLEANING YOUR EQUIPMENT
Typical cleaning instructions include disassembling all the parts and washing with warm water and dish detergent (do not wash the tubing), and then rinsing, drying with a lint-free cloth, and air drying thoroughly. It is a good idea to sterilize the nebulizer parts once a day. The easiest most efficient way is to soak the parts in a solution of 1 part white vinegar to 2 parts warm water for a period of 20 minutes, finally, rinse and air dry. However, it is a good idea to check with the manufacturer for specific cleaning instructions.
The tubing usually remains dry because air is blowing out through it. If you notice persistent moisture inside of the tubing, be sure to contact your equipment provider. Occasional moisture can be blown out with the compressor.
The compressor should also be wiped down frequently with a clean damp cloth to keep the outside clean. Follow manufacturer recommendations.
STORAGE
Equipment should always be stored in an area that is free from dust and animal hair.
SOME FINAL TIPS FROM USERS:
- Compressors that work on a car battery can be used to do a treatment while you are transporting your child to or from school or other activities.
- The same type of portable compressor could be used when going camping. Be sure to use disposable equipment. You might also consider using an MDI instead.
- When delivering a treatment to an infant or small child, the medication can be directed towards the child's nose and mouth (called the blow-by method). Some children will accept a treatment through a mask attached to the nebulizer cup.
BE GOOD TO YOURSELF
Nebulizers and compressors are an important part of continuing care for people with CF. Be sure you are providing yourself with the best care possible.
Choosing a Nebulizer
Different nebulizers have different rates of drug output, efficiency of delivery and impact on particle size, which can impact the effectiveness of a treatment. All prescription medications go through extensive clinical trials with strict controls over equipment. Thus, data on the effectiveness of a medication is based on the equipment that was used during the trials. Some drugs that have been on the market for a while have been tested with multiple nebulizers, not all of which have been found to deliver the needed dose and particle size for optimal distribution in the lungs. The newer nebulizers, such as the Sidestreamtm, are faster, but have not been found to be as effective with antibiotics. It is also important to note that faster is not always better: particle size and amount of drug delivered to the small airways are important factors in optimizing the impact of the drug on your lungs. The following is a brief description of the most common nebulizers and a table on recommended uses.
The Pari LC Plus is a reusable nebulizer, which lasts 6-12 months. The LC Plus is advertised as increasing drug delivery. The patient's breathing controls the output rate of the nebulizer by use of a valve system; therefore little medication is lost during expiration. This nebulizer has been shown in studies to be an efficient way to deliver TOBI® and most other medications.
The Pari LC Star is a resuable nebulizer, which will last 6-12 months. This nebulizer promises increased drug delivery, and a high percentage of particles of optimum size for good delivery to the lungs. The patient's breathing contols the output rate of the nebulizer by the use of a valve system and there is less medication lost during expiration than with the Pari LC Plus. The Pari LC Star has been shown to be the best way to deliver Colistin (an antibiotic prescribed to some CF patients), because it can nebulize the medication efficiently without developing foam. This nebulizer can also be used to deliver other medications.
The Invacare Sidestreamtm is a reusable nebulizer, which will last 12 months. This nebulizer is advertised as a faster way to deliver medications. It provides a high number of particles in a size range that is easily deposited in the lung. This nebulizer is now approved for use with Pulmozyme and can also be used with bronchodilators such as albuterol, ipratropium (Atroventtm), and cromolyn (Intaltm). However, it should not be used with antibiotics, like TOBI®, or amphotericin, a drug used to treat fungal infections. One disadvantage of this nebulizer is its continuous operation. Since it has no valve, the nebulizer continues to pump during exhalation and an estimated 25-50% of the medication is wasted.
Disposable or standard nebulizers available in hospitals and home care companies come in many shapes and sizes, but generally have the same features. They are designed for one-time,one patient use, but can be washed and re-used for a limited amount of time (usually one month or so). They are inexpensive and are designed to nebulize common medications like albuterol, (Intaltm) and Atroventtm. One disadvantage of this type of nebulizer is the continuous operation; because the nebulizer continues to put out mist during exhalation (instead of only during inspiration), 25-50% of the medication is wasted.
Recommended Nebulizers for Common CF Medications
Medication Pari LC Plus Pari LC Star Sidestream Disposable Albuterol Yes Yes Yes Yes Atrovent Yes Yes Yes Yes Intal Yes Yes Yes Yes Pulmozyme Yes Yes Yes Yes TOBI Yes Yes NO NO Colistin Yes Yes NO NO Amphotericin Yes Yes NO NO GI and Nutrition
- Your Digestive System and How It Works
by the National Digestive Diseases Information Clearinghouse (NIDDK)- Enzymes and How to Use Them
by Julie Matel, MS RD- GERD and CF
by Richard Moss, MD- Tube Feedings
by Julie Matel, MS RD- CF-Related Diabetes Mellitus
by by Noreen Henig, MD
Your Digestive System and How It Works
by The National Digestive Diseases Information Clearinghouse (NIDDK)
The digestive system is a series of hollow organs joined in a long, twisting tube from the mouth to the anus. Inside this tube is a lining called the mucosa. In the mouth, stomach, and small intestine, the mucosa contains tiny glands that produce juices to help digest food.
Two solid organs, the liver and the pancreas, produce digestive juices that reach the intestine through small tubes. In addition, parts of other organ systems (for instance, nerves and blood) play a major role in the digestive system.
Why Is Digestion Important?
When we eat such things as bread, meat, and vegetables, they are not in a form that the body can use as nourishment. Our food and drink must be changed into smaller molecules of nutrients before they can be absorbed into the blood and carried to cells throughout the body. Digestion is the process by which food and drink are broken down into their smallest parts so that the body can use them to build and nourish cells and to provide energy.
How Is Food Digested?
Digestion involves the mixing of food, its movement through the digestive tract, and chemical breakdown of the large molecules of food into smaller molecules. Digestion begins in the mouth, when we chew and swallow, and is completed in the small intestine. The chemical process varies somewhat for different kinds of food.
Movement of Food Through the System
The large, hollow organs of the digestive system contain muscle that enables their walls to move. The movement of organ walls can propel food and liquid and also can mix the contents within each organ. Typical movement of the esophagus, stomach, and intestine is called peristalsis. The action of peristalsis looks like an ocean wave moving through the muscle. The muscle of the organ produces a narrowing and then propels the narrowed portion slowly down the length of the organ. These waves of narrowing push the food and fluid in front of them through each hollow organ. The first major muscle movement occurs when food or liquid is swallowed. Although we are able to start swallowing by choice, once the swallow begins, it becomes involuntary and proceeds under the control of the nerves.
The esophagus is the organ into which the swallowed food is pushed. It connects the throat above with the stomach below. At the junction of the esophagus and stomach, there is a ringlike valve closing the passage between the two organs. However, as the food approaches the closed ring, the surrounding muscles relax and allow the food to pass.
The food then enters the stomach, which has three mechanical tasks to do. First, the stomach must store the swallowed food and liquid. This requires the muscle of the upper part of the stomach to relax and accept large volumes of swallowed material. The second job is to mix up the food, liquid, and digestive juice produced by the stomach. The lower part of the stomach mixes these materials by its muscle action. The third task of the stomach is to empty its contents slowly into the small intestine.
Several factors affect emptying of the stomach, including the nature of the food (mainly its fat and protein content) and the degree of muscle action of the emptying stomach and the next organ to receive the contents (the small intestine). As the food is digested in the small intestine and dissolved into the juices from the pancreas, liver, and intestine, the contents of the intestine are mixed and pushed forward to allow further digestion.
Finally, all of the digested nutrients are absorbed through the intestinal walls. The waste products of this process include undigested parts of the food, known as fiber, and older cells that have been shed from the mucosa. These materials are propelled into the colon, where they remain, usually for a day or two, until the feces are expelled by a bowel movement.
Production of Digestive Juices
The glands that act first are in the mouth--the salivary glands. Saliva produced by these glands contains an enzyme that begins to digest the starch from food into smaller molecules.
The next set of digestive glands is in the stomach lining. They produce stomach acid and an enzyme that digests protein. One of the unsolved puzzles of the digestive system is why the acid juice of the stomach does not dissolve the tissue of the stomach itself. In most people, the stomach mucosa is able to resist the juice, although food and other tissues of the body cannot.
After the stomach empties the food and juice mixture into the small intestine, the juices of two other digestive organs mix with the food to continue the process of digestion. One of these organs is the pancreas. It produces a juice that contains a wide array of enzymes to break down the carbohydrate, fat, and protein in food. Other enzymes that are active in the process come from glands in the wall of the intestine or even a part of that wall.
The liver produces yet another digestive juice--bile. The bile is stored between meals in the gallbladder. At mealtime, it is squeezed out of the gallbladder into the bile ducts to reach the intestine and mix with the fat in food. The bile acids dissolve the fat into the watery contents of the intestine, much like detergents that dissolve grease from a frying pan. After the fat is dissolved, it is digested by enzymes from the pancreas and the lining of the intestine.
Absorption and Transport of Nutrients
Digested molecules of food, as well as water and minerals from the diet, are absorbed from the cavity of the upper small intestine. Most absorbed materials cross the mucosa into the blood and are carried off in the bloodstream to other parts of the body for storage or further chemical change. As already noted, this part of the process varies with different types of nutrients.
Carbohydrates: Based on a 2,000-calorie diet, it is recommended that 55 to 60 percent of total daily calories be from carbohydrates. Some of our most common foods contain mostly carbohydrates. Examples are bread, potatoes, legumes, rice, spaghetti, fruits, and vegetables. Many of these foods contain both starch and fiber.
The digestible carbohydrates are broken into simpler molecules by enzymes in the saliva, in juice produced by the pancreas, and in the lining of the small intestine. Starch is digested in two steps: First, an enzyme in the saliva and pancreatic juice breaks the starch into molecules called maltose; then an enzyme in the lining of the small intestine (maltase) splits the maltose into glucose molecules that can be absorbed into the blood. Glucose is carried through the bloodstream to the liver, where it is stored or used to provide energy for the work of the body.
Table sugar is another carbohydrate that must be digested to be useful. An enzyme in the lining of the small intestine digests table sugar into glucose and fructose, each of which can be absorbed from the intestinal cavity into the blood. Milk contains yet another type of sugar, lactose, which is changed into absorbable molecules by an enzyme called lactase, also found in the intestinal lining.
Protein: Foods such as meat, eggs, and beans consist of giant molecules of protein that must be digested by enzymes before they can be used to build and repair body tissues. An enzyme in the juice of the stomach starts the digestion of swallowed protein. Further digestion of the protein is completed in the small intestine. Here, several enzymes from the pancreatic juice and the lining of the intestine carry out the breakdown of huge protein molecules into small molecules called amino acids. These small molecules can be absorbed from the hollow of the small intestine into the blood and then be carried to all parts of the body to build the walls and other parts of cells.
Fats: Fat molecules are a rich source of energy for the body. The first step in digestion of a fat such as butter is to dissolve it into the watery content of the intestinal cavity. The bile acids produced by the liver act as natural detergents to dissolve fat in water and allow the enzymes to break the large fat molecules into smaller molecules, some of which are fatty acids and cholesterol. The bile acids combine with the fatty acids and cholesterol and help these molecules to move into the cells of the mucosa. In these cells the small molecules are formed back into large molecules, most of which pass into vessels (called lymphatics) near the intestine. These small vessels carry the reformed fat to the veins of the chest, and the blood carries the fat to storage depots in different parts of the body.
Vitamins: Another vital part of our food that is absorbed from the small intestine is the class of chemicals called vitamins. The two different types of vitamins are classified by the fluid in which they can be dissolved: water-soluble vitamins (all the B vitamins and vitamin C) and fat-soluble vitamins (vitamins A, D, and K).
Water and salt: Most of the material absorbed from the cavity of the small intestine is water in which salt is dissolved. The salt and water come from the food and liquid we swallow and the juices secreted by the many digestive glands.
How Is the Digestive Process Controlled?
Hormone Regulators
A fascinating feature of the digestive system is that it contains its own regulators. The major hormones that control the functions of the digestive system are produced and released by cells in the mucosa of the stomach and small intestine. These hormones are released into the blood of the digestive tract, travel back to the heart and through the arteries, and return to the digestive system, where they stimulate digestive juices and cause organ movement. The hormones that control digestion are gastrin, secretin, and cholecystokinin (CCK):
Gastrin causes the stomach to produce an acid for dissolving and digesting some foods. It is also necessary for the normal growth of the lining of the stomach, small intestine, and colon.
Secretin causes the pancreas to send out a digestive juice that is rich in bicarbonate. It stimulates the stomach to produce pepsin, an enzyme that digests protein, and it also stimulates the liver to produce bile.
CCK causes the pancreas to grow and to produce the enzymes of pancreatic juice, and it causes the gallbladder to empty.
Nerve Regulators
Two types of nerves help to control the action of the digestive system. Extrinsic (outside) nerves come to the digestive organs from the unconscious part of the brain or from the spinal cord. They release a chemical called acetylcholine and another called adrenaline. Acetylcholine causes the muscle of the digestive organs to squeeze with more force and increase the "push" of food and juice through the digestive tract. Acetylcholine also causes the stomach and pancreas to produce more digestive juice. Adrenaline relaxes the muscle of the stomach and intestine and decreases the flow of blood to these organs.
Even more important, though, are the intrinsic (inside) nerves, which make up a very dense network embedded in the walls of the esophagus, stomach, small intestine, and colon. The intrinsic nerves are triggered to act when the walls of the hollow organs are stretched by food. They release many different substances that speed up or delay the movement of food and the production of juices by the digestive organs.
The National Digestive Diseases Information Clearinghouse (NDDIC) is a service of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The NIDDK is part of the National Institutes of Health under the U.S. Department of Health and Human Services. Established in 1980, the clearinghouse provides information about digestive diseases to people with digestive disorders and to their families, health care professionals, and the public. NDDIC answers inquiries, develops and distributes publications, and works closely with professional and patient organizations and Government agencies to coordinate resources about digestive diseases.
Publications produced by the clearinghouse are carefully reviewed by both NIDDK scientists and outside experts.
This e-text is not copyrighted. The clearinghouse encourages users of this e-pub to duplicate and distribute as many copies as desired.
Back to top
Enzymes and How to Use Them
by Julie Matel, MS RDAn enzyme is a protein molecule produced by the body to digest the foods we eat. Lack of enough enzymes can result in poor digestion and absorption of nutrients, cramping, diarrhea, and weight loss.
In Cystic Fibrosis mucous plugs can block the pancreas and prevent enzymes from entering the small intestine, which leads to improper digestion of foods. Without these digestive juices, the intestines cannot absorb fats and proteins completely, so nutrients pass out of the body unused. The stools become very large, lighter in color, greasy, and will float on top of the water in the toilet. Unabsorbed fats may also cause excessive intestinal "gas", an abnormal swollen belly, and abdominal pain or discomfort. Weight loss or difficulty maintaining adequate weight can occur.
Not all people with CF are pancreatic insufficient, but those who are will need to take enzymes. If your doctor has prescribed enzyme replacements, here are some suggestions for their use:
- Be sure to ask for brand name enzymes at your pharmacy. Do not accept generic enzymes. If necessary, ask your doctor to specify this by writing "Do not substitute" on the prescription.
- Take enzymes immediately before meals and snacks.
- Very large meals, or high-fat foods may require extra enzymes. Ask your doctor for dosing information.
- Foods containing mostly simple sugars may not require enzymes, if those foods are eaten alone.
Examples: sodas, jello, juice, fruit, nectars, and hard candies like Life Savers® or lemon drops. You need to take enzymes if there are 3 or more grams of fat, or 5 or more grams of protein in a food.- If meals last more than 30 minutes, you may need more enzymes. Taking enzymes several times during the meal may work better than taking them all at once.
- Don't chew, blenderize, or crush enzymes. They will be destroyed.
- Check mouth after eating for any remaining enzyme beads. They can cause mouth sores if left in the mouth too long.
- If swallowing capsules is difficult, pull them apart and sprinkle them onto a food that doesn't require chewing, such as yogurt, applesauce, juice, nectar, honey, or jelly. Don't sprinkle onto hot foods.
- If you have more than two bulky, floating, oily stools per day, or stomach aches after eating, this may indicate malabsorption and the need for increasing enzyme dosage.
- Keep enzymes in a cool dry place, but do not refrigerate.
Back to top
GERD, Propulsid, and Cystic Fibrosis
by Richard Moss, MD, Stanford CF Center DirectorGastroesophageal reflux disease (GERD) is a common complication of cystic fibrosis. It is frequently treated with Propulsid, a medication that has been in the news recently because of a decision by the manufacturer, Janssen Pharmaceuticals, to stop marketing Propulsid in the US as of July 14, 2000. This is a voluntary action taken in consultation with the FDA.
What is GERD?
Gastroesophageal reflux disease is the return flow of gastric contents into the esophagus. Normally food passes from the esophagus into the stomach and does not return. A muscle, the lower esophageal sphincter or LES, regulates the flow of food from the esophagus into the stomach. If the LES muscle relaxes when it should be tightening, food can reflux (go back up) from the stomach to the esophagus. Stomach acids can be included in this reflux, causing heartburn.
How is GERD treated?
Treatment for GERD may include
- Position changes such as sleeping with the upper part of the body propped up about 30 degrees
- Taking over- the-counter antacids like Maalox or Mylanta or low dose Zantac
- Taking prescription antacids such as high-dose Zantac , Prilosec, Prevacid, or Pepcid
- Using a pro-motility drug such as Propulsid.
What is Propulsid?
Propulsid is an oral prescription drug treatment approved by the FDA in 1993 for severe nighttime heartburn experienced by adults with GERD who were not adequately helped by other therapies. Because of its effectiveness and generally excellent side effect profile, it has been one of the most commonly prescribed drugs in the US in the last decade. Propulsid has been shown to increase the motility (movement) of the muscles in the GI tract including the esophagus, and to tighten the LES, keeping food in the stomach from refluxing into the esophagus. There are very few other medications which can do this. The major alternative medications to Propulsid, Reglan (metoclopromide) or bethanechol, are generally less effective than Propulsid. Also, they may cause side effects that are troubling, such as involuntary muscle contractions that can be quite severe.
Why is Propulsid used in CF?
GERD occurs in CF more commonly than in the general population. Besides the heartburn symptoms of GERD itself, physicians often are concerned that GERD may contribute to pulmonary symptoms. It is well established that GERD can lead to silent aspiration of stomach contents into the airways, causing pulmonary symptoms such as shortness of breath, wheezing, coughing, sputum production, bronchitis, and pneumonia. Thus, patients with CF who have GERD may receive Propulsid for GI and sometimes pulmonary symptoms.
Why use Propulsid for children?
In infants and children, Propulsid has been considered the drug choice for GERD based upon clinical studies. The 2000 United States Pharmacopeial Convention states the following: “Serious adverse effects including death have been reported in infants and children treated with cisapride. Cisapride should be used with caution and recommended doses should not be exceeded”(page 900). The recommended pediatric dose is 0.1-0.2mg/kg/dose. Total daily dose is not to exceed 1mg/kg/day.
The most common problem with Propulsid toxicity has been cardiac arrhythmias. Some, but not all, patients with this problem have had pre-existing heart disease or risk factors for arrhythmias.
Why is Propulsid being taken off the market?
Propulsid's labeling has been revised several times since 1993 to reflect concern about potential cardiac toxicity. In a treatment experience involving millions of patients, there were reports of 341 heart abnormalities possibly or probably related to Propulsid, including 80 deaths. The ages of the fatal cases, and the co-existence of known heart disease, were not reported. However, the company did state “most of these adverse events occurred in patients who were taking other medications or suffering from underlying conditions known to increase risk of cardiac arrhythmia associated with cisapride.” In CF, some other medications sometimes needed to treat CF-related complications can interact with Propulsid and increase its potential to cause cardiac arrhythmias. In particular, some antibiotics (erythromycin, clarithromycin) and antifungal agents (itraconazole) can increase Propulsid toxicity. You should never take these drugs in combination without discussing the issue with your doctor first.
What alternatives do I have ?
There are a number of options:
- Check with your doctor about whether to stop Propulsid or continue. The risk of Propulsid is reduced, but perhaps not entirely eliminated, by checking the electrocardiogram (ECG) on treatment and looking for any abnormalities in the rhythm of the heart.
- If continuing on the medication, get an ECG on Propulsid. Make sure the “QTc interval” is specifically checked, since this is the specific part of the conduction path affected. There are age-related normal ranges for this measurement.
- Non-drug measures should be reinforced including dietary changes (e.g., alcohol exaggerates GERD) and positioning.
- Antacid medications should be used.
- In cases where over-the-counter or prescription antacids are not sufficient, consider other pro-motility agents like Reglan and bethanechol. The decision should factor in their reduced efficacy and much increased (non-fatal) side effects compared to Propulsid.
- After July 14, 2000, Propulsid will continue to be available to physicians for severe cases where no alternative therapy can be used, if specific clinical eligibility criteria are met.
Back to top
Some children and adults with cystic fibrosis are not able to meet their nutritional needs for growth, weight gain or maintenance with oral intake alone. This is when supplementation with tube-feedings can be beneficial. There are several different types of tube-feedings that can be used. Two of the most common options include nasogastric and gastrostomy tubes.
What are the pros and cons for each type of tube feeding?
Nasogastric Tube
The nasogastric tube (NG) is a soft flexible tube that runs through the nose and into the stomach. Placement of an NG tube does not require surgery and can be taken out and replaced daily or left in for several days or longer. One benefit is that an individual can continue to eat by mouth with the NG tube in place.This is important for an infant who is developing sucking and swallowing skills or for an adult who would like to continue to eat by mouth for social reasons.
Putting in and taking out the tube for feedings has its drawbacks. This process can be uncomfortable and cause irritation in the nose, throat, or esophagus. Some people vomit, have reflux (backwashing of stomach contents), delayed emptying of stomach contents, or a delay in the development of feeding skills by mouth. In some cases the tube can interfere with breathing and may not be the ideal mode for a person with severe CF. And finally, some people object to its appearance because it is taped to the face when not being used for feeding. Most people, however, tolerate tube feedings via an NG tube very well once it is in place and ready for use.
Gastrostomy Tube
Unlike the NG tube, which is used for short-term feeding difficulties, a gastrostomy tube (GT) can be used for more long-term feeding or weight gain problems. A GT is placed either surgically or by a special procedure called endoscopy directly into the stomach, bypassing the mouth altogether. The GT has many advantages. Different from the NG tube, the GT can easily be concealed by clothing and does not cause throat and mouth irritation.Similar to the NG tube, a GT will allow a person to continue to eat by mouth and can be removed when no longer needed. One option of both tubes is to have feeds running throughout the night so that daytime oral intake can continue.
The disadvantages of a GT are that more equipment is required for its use, daily skin care is needed, and reflux can still occur.
Excessive weight loss can hinder growth and development in children and contribute to decreased immune function and quality of life in adults. One way to help achieve an adequate calorie diet is to supplement with tube feedings. There are quite a few options that can be individualized to meet the needs of each person with CF, big or small! Ask your medical team to help you choose the one that is right for you or your child.
Back to GI and Nutrition Topics
Cystic Fibrosis Related Diabetes Mellitus (CFRDM) is a common and serious complication of CF. Frequency increases with age, with as many as 40% of adults over age 19 and 25% of children with CF developing CFRDM. It is also distinctly different from the more common non-CF types of diabetes, which go by the names of Type I or Type II or juvenile or adult onset diabetes. Although "classic" symptoms of diabetes include increased hunger while losing weight, increased thirst, and increased urination, few if any patients with CFRDM will have these.
In CFRDM, there is too little insulin to let glucose, the "energy molecule," into the cells. Insulin is a product of the pancreas, one of the main organs affected by CF that enables sugar to be moved from the blood to the cells where it can be used or stored. Thus in diabetes, even if there is plenty of glucose in the blood, the body cannot use it for energy and must turn to other sources, like protein that has been stored in the body. This creates a situation in which weight loss is common and weight gain is almost impossible, factors most persons with CF already are challenged to manage effectively. In addition, there are detrimental effects to other organs resulting from excessive blood glucose levels, such as eye, kidney, and heart disease. The white blood cells, critical for fighting infection, also do not work well when blood sugar levels are too high. As you can guess, in persons with cystic fibrosis, undiagnosed or untreated CFRDM can contribute to weight problems, more frequent pulmonary exacerbations or other serious complications.
Making the Diagnosis
It is important for those with CF over the age of 16 years to be screened annually for the condition since the external symptoms may be subtle and are so closely associated with more typical CF symptoms. Younger children should be tested if weight gain or frequent infections are problems. Prednisone, a medication used in some patients, can cause glucose intolerance (high blood sugar), but does not promote the development of CFRDM.The diagnosis of CFRDM is made by doing a 2 hour glucose tolerance test that requires three blood tests immediately following a period of fasting. The first blood test is taken while fasting. Patients will be requested to abstain from food and all but clear liquids for at least six hours preceding the first test. Following this test, the person drinks an orange-flavored sugar solution. One hour later the second blood test is taken. The final blood draw occurs one hour after the second one. Oral glucose tolerance tests should be done when one is well, as being sick may alter the results.
Stages of CFRDM
There are three stages of CFRDM, all of which can occur without symptoms. The first stage is impaired glucose tolerance. In this stage, blood sugar is usually normal except at times of stress, as with a pulmonary exacerbation. The second stage is called CFRDM without fasting hyperglycemia (high blood sugar). In this stage, blood sugar goes too high within two hours after eating, but returns to normal between meals. The third stage is called CFRDM with fasting hyperglycemia. With this stage, blood sugar levels are always high. It is believed that patients start with Stage 1 and then progress through Stage 2 before reaching Stage 3. Although we cannot prevent someone's diabetes from becoming more severe, early identification and intervention helps prevent significant weight loss and more severe complications. In times of stress, the body may become more glucose intolerant. Thus, someone with Stage I can have features of Stage II or someone with Stage II can have high between-meal blood sugars. However, when the stress is removed, such as following successful treatment for a pulmonary exacerbation, glucose tolerance will return to normal. Studies on the impact of CFRDM on the long-term health of people with CF have been inconclusive, however we do know that maintaining weight and nutritional status are strong contributors to maintaining good health.Treatment Options
Treatment options vary by stage and symptoms, and are different from treatment of non-CF diabetes. The Stanford CF Center is now working closely with diabetes specialists in the Stanford University Department of Medicine who have significant research and clinical interests in CFRDM. Anna Simos is a diabetes educator who works closely with Julie Matel, the CF Center nutritionist. Anna is often available to come to clinic to help evaluate patients with evidence of CFRDM. One of the key points in treating CFRDM is NOT TO CUT CALORIE INTAKE. So, unlike other forms of diabetes, restricting one's diet to limit total calorie intake is not healthy and special dietary needs must be recognized and incorporated to maintain weight. Treatment varies for each individual and may include different eating strategies, insulin pills or insulin shots.CFRDM Research
The Stanford CF Center is participating in a study to identify optimum therapy for patients with CFRDM without fasting high blood sugar (Stage 2). If you are diagnosed with this stage of CFRDM, you may be interested in participating in the study. If you wish to learn more about CFRDM, ask your doctor.
Back to GI and Nutrition Topics
CF and Bone Health
- C.F. and Osteoporosis
by Laura Bachrach, M.D.- Calcium and Bone Health
by Julie Matel, MS RD
C.F. and Osteoporosis
by Laura Bachrach, M.D.Bone Weakness in Patients with Cystic Fibrosis
Why? Who? Treatment?There is evidence that patients with cystic fibrosis face an increased risk of bone problems. Deficits in bone strength may be mild, a condition called "osteopenia" in which the bone mineral density is low for age but there are no symptoms. In other cases, patients have developed "osteoporosis", with very low bone mineral and loss of bone strength. Individuals with osteoporosis may have fractures of the spine, hip, rib or other sites with minimal or no trauma. These fractures are often quite painful and may result in permanent deformities such as a "hump" in the spine.
There is no single "cause" of bone problems in cystic fibrosis. Instead the problems arise because of inadequate nutrition, inactivity, sex hormone deficiency, and glucocorticoid (steroid) use. Because CF patients experience fat malabsorption, they may be underweight and may develop low levels of vitamin D. Maintaining a healthy weight and adequate vitamin D is important for developing and keeping bones strong. Because of pulmonary disease, CF patients may limit exercise and even require bed rest for periods of time. Immobility increases the risk of bone loss. Puberty may begin late in CF and many adults experience declining levels of their sex hormones (estrogen and testosterone). Finally, chronic use of glucocorticoids (such as prednisone) may cause bone loss.
Childhood and adolescence are particularly critical periods for establishing bone health. By age 30, adults have achieved their peak bone mass, which serves as the "bone bank" for life. If gains in bone strength during childhood and adolescence fall short, there is an increased risk of early osteoporosis. The risk factors described above may interfere with the normal gains in bone strength or may contribute to early bone loss. The amount of bone mineral in your skeleton can be estimated using a technique called dual energy x-ray absorptiometry (DXA). This test is painless and has far less radiation than standard x-rays. DXA studies have shown that CF patients of all ages may develop osteopenia and osteoporosis. For this reason, it is important to do as much as possible to foster your bone health beginning in childhood.
To prevent and treat these bone problems, it is important to address all of the risk factors mentioned above. CF patients should try to maintain a healthy body weight and should consume the recommended amount of bone-building calcium and vitamin D. The recommended daily calcium intake is 800 milligrams for ages 4-8, 1300 milligrams for ages 9-18, and 1000 milligrams for adults. An eight ounce cup of milk or yogurt contains about 300 milligrams of calcium. The best source of calcium is food, but supplements (pills) can be added if necessary to insure that the goal is met. A blood test (called a 25-hydroxyvitamin D level) can determine if your vitamin D stores are adequate. Sometimes, CF patients require more than 800 I.U. of vitamin D to maintain stores of this important vitamin. Weight-bearing exercise (such as jogging, dancing, or weight lifting) is the best for bone building if tolerated. Even standing and gentle walking during illness may help prevent the bone loss that occurs with bed rest. Your doctor can help you determine if your sex steroid levels are adequate or whether you might benefit from added estrogen or testosterone treatment. If you require glucocorticoid treatment for your lung disease, you and your doctor should work to reduce to the lowest dose that controls your breathing symptoms. Inhaled steroids may cause less bone loss than steroid pills. Your bone health can be monitored by DXA. It may be reasonable to perform a DXA test during adolescence to establish a baseline. Certainly, the test is appropriate in any patient with a history of fracture. It is also important to monitor patients in the first several months after lung transplantation, when bone loss may be very rapid.
For some patients, these general measures will be sufficient to prevent bone problems. For others, osteoporosis may develop despite attention to diet, exercise and hormones. These patients may experience fractures. There are several medications on the market to treat osteoporosis. Unfortunately, they have been tested mostly in women with post-menopausal osteoporosis. A drug called pamidronate has been used successfully to reduce weakened bones after transplantation in CF patients. It has also been tried in a handful of patients with osteoporosis unrelated to tranplantation. Unfortunately, some of these individuals have complained of severe bone pain and other side effects following this therapy. Further research is needed to determine if they are safe and effective in treating osteoporosis in cystic fibrosis.
To learn more about CF and osteoporosis, here is a list of some recent articles on the topic:
Aries RM, Neuringer IP, Weiner MA, Egan TM, Ontjes D. Severe osteoporosis before and after lung transplantation. Chest 1996; 109: 1176-1183.
Bhudhikanok GS, Wang M-C, Marcus R, Harkins A, Moss RB, Bachrach LK. Bone acquisition and loss in children and adults with cystic fibrosis: a longitudinal study. J Pediatr 1998; 133: 18-27.
Henderson RC, Madsen CD. Bone density in children and adolescents with cystic fibrosis. J Pediatrics 1996; 128: 28-34.
Laursen EM, Molgaard C, Michaelsen KF, Koch C, Miller J. Bone mineral status in 134 patients with cystic fibrosis. Arch Dis Child 1999; 81: 235-249.
Mortensen LA, Chan GM, Alder SC, Marshall BC. Bone mineral status in prepubertal children with cystic fibrosis. J Pediatr 2000; 136: 648-652.
Back to CF and Bone Health Topics
Calcium is an important mineral for building strong bones. Making sure you or your child gets enough calcium in the diet is one step you can take to help prevent osteoporosis, a debilitating bone disease.Are you eating calcium rich foods?
The following are groups of foods that contain high and medium amounts of calcium. It takes 3 servings from the medium-calcium foods to equal 1 serving from the high-calcium foods.
HIGH-CALCIUM FOODS
Serving Size = 1 cup milk, yogurt, pudding;
or 1 1/2 oz cheese
Contains approximately
300 mg calcium per servingMEDIUM-CALCIUM FOODS
Serving Size = ˝ cup
Contains approximately
75-150 mg calcium per servingwhole, lowfat or nonfat milk or chocolate milk
nonfat or lowfat yogurt
milkshake
string cheese
cheese
pudding or custard
Calcium-enriched orange juice1/8 of a 12" pizza
cottage cheese (lowfat or nonfat)
frozen yogurt or ice cream
tofu (calcium-set)
corn tortillas (2)
broccoli
refried beans
almonds (1/4 cup)How much calcium do you or your child need?
Age
Calcium Needed
(milligrams)Daily High-Calcium
Food Servings NeededChildren
1 to 5 years
6 to 10 years
800
800-1200
3
3 to 4Teens/Young Adults
11 to 24 years
1200-1500
4Adults
25 to 65 years
1000
3What if I am lactose intolerant and can't digest dairy products?
Lactose is the sugar present in milk. Some people make less of the enzyme needed to break down lactose for absorption into the bloodstream. The lactose then ends up in the large intestine where bacteria break it down and produce gas. This can cause gastrointestinal upset and bloating.
There are Lactaid© pills you can take right before consuming dairy products that breakdown the lactose for you! Lactaid© milk, which has been treated with the lactose enzyme, is also available in most supermarkets.
Eating on the Run?
When eating at fast-food restaurants…
- choose cheese burgers instead of plain hamburgers
- order pasta or pizza with cheese and vegetables
- choose milk or milkshakes instead of soda
- opt for the salad bar and choose vegetables and beans topped with shredded cheese and almonds
- choose a bean and cheese burrito or a couple of tacos
Why bother eating all these foods, when I can just take a calcium supplement?
Calcium-rich foods are packaged with an important combination of nutrients, like vitamin D and protein, that you won't find in a supplement alone. These other nutrients are important in that they increase the absorption of calcium.
If you do take calcium supplements, choose a supplement that provides about 100% of the recommended dietary intake for calcium and take them in divided doses with meals. Avoid oyster shell derived calcium due to the high levels of lead and other heavy metals that they may contain, which can be toxic.
Back to CF and Bone Health Topics
The current mean life expectancy for someone with cystic fibrosis is 32 years old. What is truly profound about this is that 32 years ago, the life expectancy was closer to 10 years. This reflects greater improvement in understanding and rapid advancements in treatment of CF, even though there is no cure yet. It also illuminates how important it is to transition care of CF at the age of 18. No longer will a 22 year just be an “old” kid with CF. That 22 year old has many, many years to live and to live well.
New Adult Center at Stanford
In recognition of the growing population of adults with cystic fibrosis, the LPCH/Stanford CF Center is working hard to develop a dedicated Adult CF Clinic. In September, an adult pulmonologist with experience at another dedicated Adult CF Clinic was hired to spearhead the endeavor. This Spring, a nurse coordinator dedicated to the Adult Clinic will be hired. Nutrition services, respiratory care services, and social work services will be readily available and there will be ongoing education for the entire clinic staff toward the care of adults.
How is adult care different?
Why all this energy toward developing an Adult CF Center? It is well known that the care of adults is different than the care of children. Although many care issues are the same, there are many that are not. In addition, there are issues that are pertinent to all adults, not just those with CF, that need to be addressed. The following is a short list of some of those issues:
Changes that can help
As someone with CF transitions from Pediatric to Adult care, it is important to reevaluate all aspects of care. Education is a top priority. Certainly, as one gets older, learning about different aspects of CF takes on new priorities. Similarly, things that were told to you as a child may mean something new now. Also, the information itself is changing so quickly that it is important to keep everyone up-to-date. Medications are re-evaluated. Specific medications that you may have been on for years and years may not have the same effectiveness as you grow up. There are many newer medications that can be used. Tailoring medical regimes to an adult life style is also important. For instance, how many of you use Flutter on your commute?
Shifting goals
Goals of care are also reevaluated. The usual goal of CF care is to preserve lung function and nutrition. There are some patients, especially those with severely compromised lung function, for whom the care needs to shift from prevention of deterioration to comfort and maximizing quality of life. This is accomplished in many different ways and is patient specific. Again, focusing on adult care is an important way to accomplish this.
Adult CF Care is not a new idea, just new to the Stanford community. The physicians and staff are looking forward to developing it to meet the needs of our community. As the changes are being made, we welcome your comments.
Back to Treating Cystic Fibrosis
The potential for using genes themselves to treat disease--known as gene therapy--is an exciting application of DNA science. Although still in its infancy, this rapidly developing field holds great potential for treating or even curing genetic and acquired diseases, using normal genes to replace or supplement a defective gene. The enormous potential of gene therapy is easily understood, but long term benefits of gene therapy are still unknown, and much research remains to be done.
Gene Therapy and CF
The starting point for gene therapy is the identification of the gene which causes a specific disease. The Cystic Fibrosis gene was discovered in 1989. CF is caused by mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Since the discovery of the CF gene, most of the work in CF gene therapy has focused on correcting the genetic defect in the lungs.
What Happens?
Researchers began replacing damaged genes about 10 years ago. One of the biggest challenges has been to find a safe delivery vehicle to carry the new genes: one that can carry sufficient quantity of genes, and can target specific parts of the body. Viruses have evolved a way of encapsulating and delivering their genes to human cells in a pathogenic manner (think of the last cold you had). So scientists have tried to take advantage of the virus's biology and manipulate its genome to remove the disease-causing genes and insert therapeutic genes. A leading contender, and one that has been used in gene therapy trials at Stanford, is an adeno-associated virus, known as AAV. In a Phase One Clinical Trial, using this virus to carry the corrected version of the CF gene, an aerosolized administration showed an efficient transfer of the gene.
What Next?
This was really good news, but once the gene is in the cell, it needs to operate correctly. Patients' bodies may reject treatments. And there is the need to regulate gene expression. All of these problems and potential problems need further study. Because of the potential for developing new medications, pharmaceutical companies are very interested in the exploration of gene therapy, and so studies are being funded. But preliminary gene therapy efforts sometimes receive too much consumer attention. Progress is being made, but there is much more research that needs to be done. Phase Two Clinical Trials of the aerosol administration of the CFTR gene are starting, but the message at this time must be, “Gene therapy is still far from clinical reality.”
See related article on The Human Genome
Back to Treating Cystic Fibrosis