
The Basics of Huntington’s Disease (Text and Audio)
Click on the link below to hear an audio recording of this article:
The Basics of Huntington’s Disease
Huntington’s disease (often abbreviated “HD”) was first described in medical literature in 1872 by Dr. George Huntington, a physician from Long Island, New York. The disease affects men and women alike, occurring at a rate of about one in every 10,000 in most Western countries. People with HD need dedicated care and support from their loved ones, which makes the number of lives touched by the disease even greater.
The age of onset of Huntington’s disease is normally between 30 and 50 years old, although there is also a form of HD that affects children and teenagers. People with HD may express a wide variety of symptoms, which physicians typically group into three categories: movement, cognitive, and psychiatric symptoms.
- Some of the movement symptoms of HD include muscle spasms, tics, rigidity, falling down, difficulty physically producing speech, and, in the later stages of the disease, difficulty swallowing (which can lead to significant weight loss). Uncontrollable movements such as writhing and twisting are also quite common symptoms of HD. Physicians sometimes refer to these uncontrollable movements as “chorea”.
- The most significant cognitive symptoms of HD are the altered organization and generally slowed processing of information in the brain. These symptoms can lead to difficulty learning new things, difficulty planning and prioritizing, impairment of one’s perception of space (where one is in relation to tables, walls, etc.), and difficulty “multitasking” (paying attention to several things at once). Individuals frequently adhere to common routines because these routines are the easiest for them to accomplish. Finally, because they have trouble organizing incoming and outgoing words in their brains, many people with HD experience difficulty communicating with others.
- Depression is the most common of the psychiatric symptoms of HD. Other symptoms include personality changes, apathy, anxiety, irritability, obsession with certain activities (such as hand washing), delirium, and mania. Denial of having HD is also a common symptom of the disease.
Sadly, somewhere between 10 and 25 years after symptoms first appear, HD usually takes such a toll on individuals that they die of pneumonia, heart failure, or other complications.
HD causes deterioration of the nerve cells in the brain, prompting significant changes in one’s ability to think, feel, and move. The cause of these symptoms remained a mystery for quite some time until doctors noticed that the disease “ran in families” and suspected its hereditary basis. The inheritance of HD (like other hereditary traits) is now known to depend upon a “chemical code” of information contained in a substance called deoxyribonucleic acid, or DNA, which exists within living cells. Understanding a bit about this chemical code helps to give better insight into the causes of HD and into treatments that may one day lead to its cure.

The chemical code of DNA is a lot like the English language: both use specific letters in a specific order to communicate specific things. But while the English language has 26 letters, the DNA code only has four–A, C, G, and T (which stand for chemical subunits of DNA). Also, while English words can consist of either a few or many letters, DNA “words” are always three letters long. In the study of genetics, these three-letter “words” are called codons. Aptly named, codons code for the future building that goes on in the nerve cell. They are a bit like blueprints. Consider this example: when a passage contains the letters C-A-T in English, this paints a picture of our favorite lazy pet. In much the same manner, when the code of DNA contains the letters G-G-C, this tells the cell to build with proline, an amino acid. For more about DNA, click here.
If codons are like blueprints, then we can think of the amino acids that result from them as unique building blocks. When these blocks are put together chemically, they create a structure known as a protein. Like buildings in modern society, proteins are where the work of the nerve cell gets done. Proteins have many different jobs: they help the cell maintain its structure, produce energy, and communicate with other cells. If it were not for the body’s millions of proteins, life as we know it could not occur.

The specific actions of a protein are determined by its unique 3-dimensional shape. This shape controls how the protein can “fit in” and interact with other parts of the cell. The shape is determined by the type of amino acids that compose the protein, as well as by the specific order they are in. Thus, as with any well-engineered building, a successfully functioning protein starts with the “blueprints” (codons).
All human cells contain a protein called huntingtin. (Please note that although “Huntington’s disease” is spelled with an “o”, the correct spelling of the protein involved is “huntingtin” with an “i.”) Although scientists have yet to determine huntingtin’s exact function, it seems to play a critical role in the events that help nerve cells function effectively. Like many other proteins, huntingtin contains within it the amino acid glutamine. In people with HD, however, there is an excess number of glutamines in a particular segment of the protein. These extra glutamines come from having too many copies of the corresponding codon (the one that codes for glutamine) in the chemical code of DNA. That codon has the letters C-A-G. In a very real sense, HD results from having too many copies of C-A-G in the DNA that codes for huntingtin protein. That is why HD is often referred to as a trinucleotide repeat disorder (“trinucleotide” being a fancy word for codon).

Exactly how many copies of C-A-G are too many? A great deal of research has been done in this area and there are many different opinions throughout the scientific literature in answer to this question. Rough estimates are as follows: People with 10 to about 35 copies of C-A-G have a normally functioning form of the huntingtin protein. Those with 40 or more have the altered huntingtin and will eventually develop symptoms of HD. For people who have 36 to 39 copies of C-A-G, the outcome is less clear. Some will develop the symptoms of Huntington’s disease and some will not. To learn more about how HD is passed on through generations, click here:
To summarize the above, Huntington’s disease is caused by too many copies of the codon C-A-G in human DNA, which puts too many copies of glutamine in the huntingtin protein. But exactly how is the altered huntingtin damaging? Unfortunately, despite valiant efforts by researchers, a definite answer to this question has yet to be found. Since the shape of the protein determines its interactions with other parts of the cell (as we learned earlier), much of the research to this point has sought to understand exactly how a shape alteration affects huntingtin’s interactions with the other components of the cell. One study suggests that the overabundance of glutamines in huntingtin causes rigid groupings of proteins. Since the components of the nerve cell are accustomed to a more flexible environment, they cannot work under the increased rigidity. The end result is basically early cell death of the nerve cell through a process called apoptosis. Another recent study suggests that the altered (and larger-than-normal) huntingtin “kidnaps” smaller proteins in the nerve cell, keeping them from doing their jobs. In this way, the altered huntingtin could indirectly damage the nerve cell. (For more information about altered huntingtin protein, click here.)
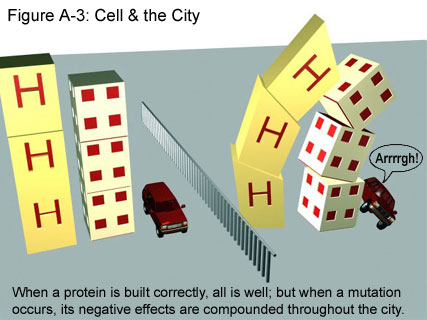
While scientists continue to work out the fine details of HD, the basic mechanism is clear. Continuing with our construction analogy, what happens when huntingtin is made in the altered form is that the “building” (the protein) does not have the specific size and shape that it was meant to have, and thus cannot function correctly in the “metropolis” that is the nerve cell. When it cannot function correctly, it hinders the action of other proteins that depend on it. The end result is a snowball effect, where the problems are continually compounded and the nerve cell becomes more and more damaged. Ultimately, after enough damage occurs, the nerve cell dies. When many other nerve cells follow suit, the problems of thinking, feeling, and moving that are associated with HD can result. For more information on nerve cells and how their deaths relate to the symptoms of HD, click here:
To leave feedback about this section of the HOPES website, please click here. Make sure to specify which article you’re referring to.
Further reading:
- Australia’s “Cooperative Research Centre for Discovery of Genes for Common Human Diseases” (Gene CRC) Web site has some fabulous tutorials at various levels of understanding.
- Cummings, C. J and Zoghbi, H.Y. “Trinucleotide Repeats: Mechanisms and Pathophysiology.” Annu. Rev. Genomics Hum. Genet. 2000. 1:281-328.
A fairly technical chapter explaining the symptoms of HD, as well as a breakdown of the number of CAG repeats in people with and without the disease. Also discussed are theories regarding the function of the altered huntingtin protein. - Falush D, et al. “Measurement of mutational flow implies both a high new-mutation rate for Huntington disease and substantial under ascertainment of late-onset cases.” Am J Hum Genet 68 (2) 2001 Feb: 373-385.
A technical analysis of the number of CAG repeats that is believed to determine whether one will develop the symptoms of HD. - “Huntington’s Disease”. Online Mendelian Inheritance in Man.
A compilation of abstracts from a multitude of different studies on HD. From case studies regarding inheritance to new methods of diagnosing HD, this is an excellent site for all the various types of HD research going on today. - “Huntington’s Disease”. Web MD.
A very reader-friendly overview of HD. Explains generally the history, symptoms, and treatment of the disease. - Li SH, Lam S, Cheng AL, Li XJ. “Intranuclear huntingtin increases the expression of caspase-1 and induces apoptosis.” Human Molecular Genetics, 2000, Vol. 9, No. 19: 2859-2867.
A highly technical paper regarding the manner in which altered huntingtin protein may cause early death (apoptosis) of the nerve cell. - Nucifora, Frederick C. Jr., et al. “Intranuclear huntingtin increases the expression of caspase-1 and induces apoptosis.” Human Molecular Genetics, 2000, Vol. 9, No. 19: 2859-2867.
Another highly technical paper regarding the manner in which altered huntingtin protein may cause early death (apoptosis) of the nerve cell.
-M. Stenerson, 7-15-03
Podcast: Play in new window | Download