 Mad Cows,
Mad Cows,
 Kuru
Kuru
 and
You!
and
You!
Introduction:
What
are prions? Originally thought to be viral mediators of disease that were
described as transmissible encephalopathies, spongiform encephalopathies
and slow virus diseases, prions, (proteinaceous
infectious
particles) have been determined to be misfolded, protease resistant proteins
which can mediate transmission of disease.
Prions have been implicated in a quartet of human diseases, specifically
kuru,
Creutzfeldt-Jakob
disease,(CJD), Gerstmann-Straussler-Scheinker, (GSS) disease,
and fatal familial insomnia, (FFI). In sheep, scrapie is believed
to be caused by prions and Bovine Spongiform Encephalopathy is a prion
disease in cattle.
Stanley
Prusiner won the
Nobel Prize in 1997 for first proposing the remarkable hypothesis
that
these prion diseases were caused by misfolded proteins, and furthermore,
elucidating the gene and a mechanism by which the misfolded wild type protein
might bring about the amyloid plaques observed clinically. The seminal
paper in which some of this information was published is available here.
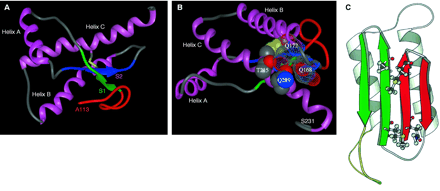
This is the third figure from the report, Molecular Biology of prion diseases. Prusiner, SB, Science 1991; 252; 1515-1522. Figure 3a depicts the structure of PrPc or the wild type PrP prion protein derived from NMR data. Figure 3c is the expected structure of the PrPSc, the misfolded, protease resistant, infectious disease causing molecule.
History:
1738: First clinical manifestation
of scrapie described.
1955: Vincent Zigas begins
clinical study of kuru in Papua New Guinea
1956: D. Carleton Gajdusek
begins investigation of kuru.
1959: WJ Hallow observes
the similarity between kuru and scrapie.
1962: H.B. Parry believed
that scrapie can be eradicated by breeding methods.
1965: First chimpanzees
injected with brain extracts of both kuru and CJD patients developed similar
symptoms to
the respective diseases.
1982: Stanley Prusiner et
al: develop animal model for studying prion infectivity.
1986: BSE epidemic in England.
Believed to arise from contaminated feed.
1988: N. Hunter observes
fibrils in BSE infected cows that are similar to scrapie protein.
1990: J. Hope determines
two alleles of protein gene linked to scrapie in sheep.
1991: Stanley Prusiner elucidates
the molecular biology of prion proteins.
1993: T.G.F. Esmonde determines
possible links to CJD caused from BSE.
1997: Stanley Prusiner wins
Nobel Prize for work in prion concept.
Scrapie has been known for as long as man has been herding sheep with the first clinical manifestation described in 1738. H.B. Parry, believed that scrapie was a genetic disease and could be eradicated with appropriate breeding controls. Parry H.B., Scrapie: a transmissible and hereditary disease of sheep. Heredity 1962; 17:75-105. He also believed that transmission by inoculation to be of importance primarily for laboratory studies but not for communicable infection in nature. Studies of the open reading frame of the PrP gene in Suffolk sheep in American have determined argued that susceptibility in Suffox sheep to scrapie is governed by the PrP codon 171 polymorphism. Goldmann, W., Hunter N., Foster, J.D., Salbaum J.M., Beyreuther K., Hope J., Two alleles of a neural protein gene linked to scrapie in sheep. Proc Natl Acad Sci USA 1990;87:2476-2480. There have been no epidemics of scrapie in sheep, however, natural scrapie is readily spread within flocks.
Unlike scrapie,
in 1986 there was and epidemic of a previously unknown disease that developed
in herds of cattle in England. Originally called Bovine Spongiform Encephalopathy
but it is commonly called mad cow disease. BSE was shown to be a prion
disease by elucidation of protease resistant PrP, (prion) protein in brains
of ill cattle. Hope, J., Reekie, L.J.D., Hunter, N., Fibrils from brains
of cow the new cattle disease containing scrapie associated protein. Nature
1988;336:390-392.
Based
upon epidemiological evidence, the BSE epidemic in England is proposed
to have been derived from the process by which MBM, a nutritional supplement,
is made. Specifically, the rendering process by which lipid rich fractions
are isolated from sheep and cattle offal might protect misfolded scrapie
prions from the sheep offal from being inactivated by the steam used in
the rendering process. Since 1988, the use of dietary protein supplements
derived from sheep and cattle offal has been prohibited in the UK, however,
it is unknown whether or not this will actually affect the incident of
BSE in England as nearly half of the BSE cases in the UK have occurred
in herds where only one cow is affected, and multiple cases of BSE in single
herds is infrequent.
Wilesmith J.W., Wells, G.A.H., Cranwell, M.P.,
Ryan, J.B.M. Bovine spongiform encephalopathy: epidemiological studies.
Vet Rec 1988; 123:638-644.
Other
lines of evidence against the possibility that the BSE epidemic might have
been caused by scrapie prions in the MBM feed are that bovine PrP differ
from sheep PrP at seven or eight residues. It has not yet been established
if scrapie prions transferred into cattle brains can initiate infection.
Goldman,W.,
Hunter, N., Martin, T., Dawson, M., Hope, J. Different forms of the PrP
genes have five or six copies of a short, G-C rich element within the protein
coding exon. J Gen Virol 1991;72:201-204.
It is
still unknown whether or not scrapie, initially implicated in CJD, or BSE
prion can cause disease in humans, although two farmers with BSE afflicted
cattle have died of CJD in 1993. Sawcer, S.J., Yuill, G.M., Esmonde,
T.G.F., et al. Creutzfeldt-Jakob disease in an individual occupationally
exposed to BSE. Lancet 1993;341:642.
Kuru and other Human Prion Diseases:
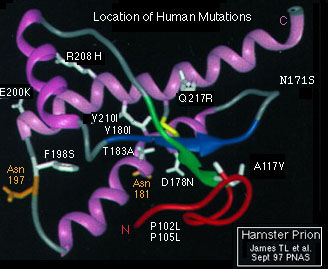
Both images from Sperling Biomedical Foundation
page. Figure 1, the native confomer predicted from NMR spectra data and
the proposed PrPSc human prion protein. Figure 2 shows the human
mutations associated with all human prion diseases superimposed on the
hamster PrPc prion gene product.
Figure 1
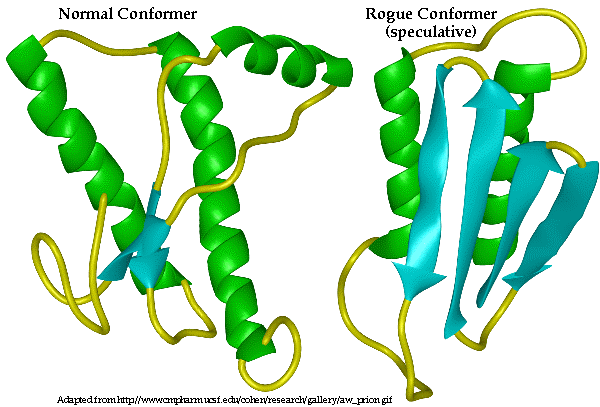
Figure 2
Kuru was first
seen by a foreigner to Papua New Guinea in 1954 by an Australian patrol
officer who observed a young Fore tribe girl who was shaking violently
and her head was jerking savagely from side to side. In 1955 Vincent Zigas,
a district medical officer in the Fore tribe region began official medical
study. However, after conventional methods for isolating the causative
agent failed, Dr. Zigas discontinued his research. Subsequently in 1956,
a researcher from Harvard, D. Carleton Gajdusek set off for Papua New Guinea
to study kuru. Dr. Gajdusek originally considered viral menigoenphalitis
but there where no clinical signs or symptoms to support this. He then
considered an environmental toxin but found no evidence. Upon assessment
of geological incident of disease, he isolated a region, approximately
35 by 25 miles across where the highest incident of disease occurred. Furthermore,
Fore women from this area, who married into other tribes carried the disease
with them, with their children having a higher prevalence of the kuru disease.
Dr. Gajdusek then considered a possible genetic linkage for predilection
to the disease. However, the tools of the day made it impossible for Dr.
Gajdusek to continue his investigations.
Then in 1959
a veterinary surgeon WJ Hallow commented in Lancet upon the similarities
between kuru and sheep scrapie. Dr. Gajdusek returned to Papua New Guinea
and sent necropsy samples to Bethesda to be intracranially inoculated into
chimpanzees. In 1965 the first batch of chimpanzees from this study developed
symptoms and signs similar to kuru.
However, two
anthropologist, Robert and Shirley Glasse determined that kuru was only
50 years old. Also, cannibalism had been uncommon in the Fore people until
1915 where they began to eat human flesh at a Kamano feast. From this point
cannibalism began to be associated with the funeral rituals. After putrefying
for three or four days, the body was baked and totally consumed. The mother
and brother ate the brains while the other men of the tribe typically forewent
the meal as they superstitiously believed it would hamper their fighting
ability. With the cessation of cannibalism, the incident of kuru significantly
decreased in the Fore people.
At the same
time as the kuru innoculated chimpanzees developed clinical signs of kuru,
other chimpanzees were inoculated with brain extracts from diseased CJD
patients. These chimps also developed CJD signs and symptoms. With the
pathological similarity of kuru, scrapie and CJD, and transfere of the
agent to animal models, multiple groups began to search for the infectious
agent.
In 1982 Stanley
Prusiner developed a Syrian hamster model for studying the infectivity
and onset of disease of prion proteins and mutations. Prusiner, S.B.,
Cochran, S.P., Groth, D.F., Downey, D.E., Bowman, K.A., Martinez, H.M.
Measurement of the scrapie agent using an incubation time interval assay.
Ann Neurol 1982;11:353-358. From this assay and subsequent elucidation
of the prion gene continuing work has been aimed at elucidating the precise
pathology of prion diseases.
Initially the
prion gene knockout mice were without abnormal phenotypes. Lipp, H.P.,
Stagliar-Bozicevic, M., Fischer, M., Wolfer, D.P., A 2-year longitudinal
study of swimming navigation in mice devoid of prion protein: no evidence
for neurological anomilies or spatial learning impairments. Behavioral
Brain Research; 1998 Sept. 95(1):47-54. However further experiments
showed increased susceptibility to prion diseases with specific prion gene
deletions or mutations. In addition, specific perceptual deficiencies and
lesions in the brain have been elucidated in the prion knockout mice. Fischer,
M., Rulicke, T., Raeber, A., Sailer, A., Moser, M., Oesch, B., Brandner,
S., Aguzzi, A., Weissman, C., Prion protein (PrP) with amino proximal
deletions restores susceptibility of PrP in mice to scrapie. EMBO J. 1996,
Mar. 15 15(5):1255-64. Shmerling, D., Heggi, I., Fischer, M., Blattler,
T., Brandner, S., Gotz, J., Rulicke, T., Flechsig, E., Cozzio, A., von
Mering, Expression of amino terminally truncated PrP in the mouse leading
to ataxia and specific cerebellar lesions, Cell, 1998 Apr. 17 93(2)203-214.
As prions do
not contain nucleic acids, they are clearly not viruses. The precise nature
of their infectivity is still some matter of dispute. However, it would
appear from work already cited, that the specific mutations in the prion
gene permits the folding of a new conformation that is protease resistant.
This new conformation then has the capacity over time to associate with
other misfolded proteins. The kinetics of these associations predict the
interactions are not cooperative, this is to say that the misfolded prion
protein does not actively recruit other misfolded prion gene products for
form multimers. The data seems rather to define a slow process by which
the occasionally misfolded prion protein will exist in the cell for an
extended period of time, after which another randomly misfolded prion protein
will associate with the first. Subsequently, as multimers of the misfolded
prion proteins associate, they have a greater statistical chance of associating
with other misfolded prion proteins. It is unknown as to whether or not
the large oligomers of the misfolded prion can promote misfolding of normally
expressed prion products. Although there is no evidence yet for this activity,
it is still an attractive hypothesis. Subsequently, large multimers or
amyloid plaques may form that may cause lesions or other cellular damage.
In human CJD, amyloid plaques are a hallmark of progressive disease whereas
in sheep and cows neural tissue that is full of holes like a sponge are
more often found.
Interestingly,
inherited CJD have mutations that may reflect the spontaneously occurring
mutations in the non inherited disease. However, these inherited mutations
do not seem in decrease the time course of the disease, just increase the
susceptibility/probability of acquiring the disease.
At this time multiple labs, who's work I've cited and partially described are actively investigating the nature of prion diseases and the mechanisms by which misfolding may increase the propensity to form plaques, or cause specific neural tissue destruction.
Some of the current labs investigating prions:
Prusiner, S.B.;
Knock-outs in mice, kinetics of disease, and mechanisms for disease progression.
Weissman C.;
Specific mutations in prion gene that effect kinetics and susceptibility
to disease.
Von Mering A,;
Mutations that bring about specific types of lesions or disease.
Wolfer D.P.;
Neural abnormalities in prion knock-out models.
Page Designer: John B. Mumm in Dr. Robert Siegels' Humans and Viruses Class. Works cited are in italics. I also made significant use of Fields Virology, Third Edition, Chapter 3, Robert A. Lamb, Robert M. Krug. Orthomyxovirdae: The Viruses and Their Replication.